
17α-羟化酶缺陷症诊治研究进展
2020-11-19 08:56:38马婧杜雅丽权金星
国际内分泌代谢杂志 2020年5期
马婧 杜雅丽 权金星
1甘肃省人民医院内分泌代谢科,兰州 730000; 2华中科技大学同济医学院附属协和医院眼科,武汉 430000
先天性肾上腺皮质增生症(CAH)是一组由肾上腺皮质类固醇合成通路各阶段催化酶的缺陷,引起以皮质类固醇合成障碍为主的常染色体隐性遗传性疾病[1]。21 羟化酶缺陷症(21OHD)是最常见的类型,占90%~95%[2]。17α-羟化酶缺陷症(17OHD)是由于CYP17A1基因突变所致的罕见CAH,该病早期无典型的临床表现,患者常以高血压、低钾血症及性发育异常就诊,88%的患者直至青春期才被确诊[3],临床上漏诊率和误诊率高[4]。
1 流行病学
17OHD的全球发病率大约为1∶50 000,约占CAH病例的1%[5]。不同地区的发病率因种族和地理位置而异,17OHD在巴西[6]、中国[7]是第二常见的CAH。Biglieri等[8]于1966年首次对该疾病进行报道。目前全球约有240余例17OHD个案被报道[5]。
2 病因和发病机制
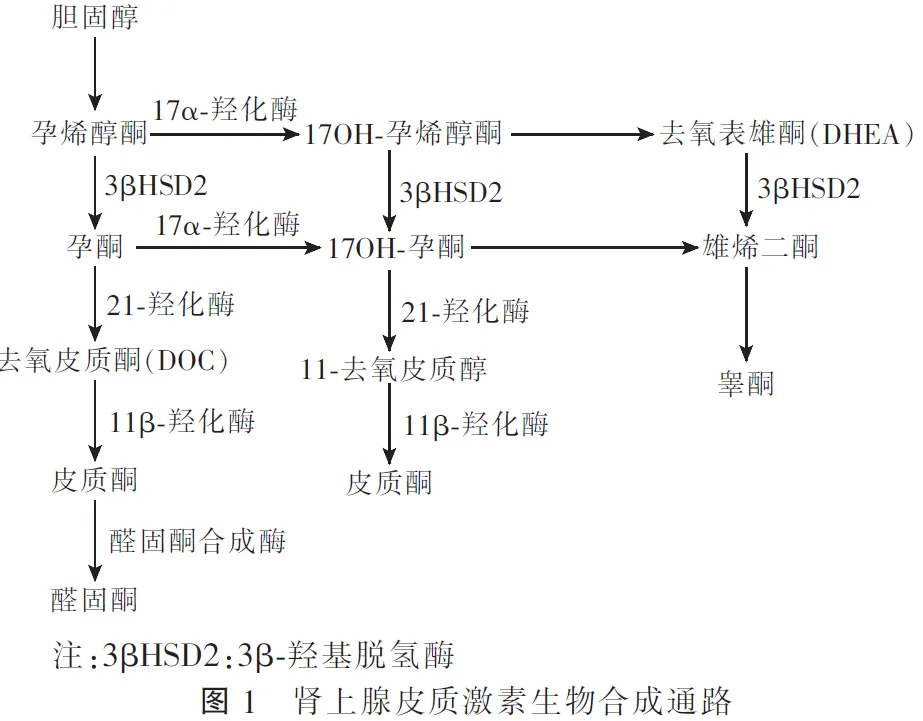
17OHD是CYP17A1基因突变所致的罕见常染色体隐性遗传病,该基因位于10q24.3,包含8个外显子和7个内含子,表达于肾上腺和性腺[9]。CYP17A1基因编码形成的P450c17酶负责催化肾上腺类固醇激素的合成,同时具有17α-羟化酶/17,20裂解酶活性:其中17α-羟化酶能够将孕烯醇酮和孕酮转化为17α-羟孕烯醇酮和17α-羟孕酮,17,20裂解酶可使17,20位碳链裂解,形成雌激素前体即去氢表雄酮和雄烯二酮(图1)。17OHD患者因CYP17A1基因突变导致部分或全部P450c17酶活性丧失,引起皮质醇和性激素合成障碍[10]。进而反馈性地刺激下丘脑和垂体分泌大量的促肾上腺皮质激素(ACTH)、卵泡刺激素(FSH)、黄体生成素(LH),引起肾上腺皮质增生并通过非17α-羟基化途径刺激11-去氧皮质酮(DOC)的过量产生,其作为醛固酮的前体物质具有强大的盐皮质激素活性,使得机体出现高血压、低钾血症、血浆肾素活性抑制,17,20-裂解酶活性的丧失会阻止性类固醇的合成,导致性幼稚。研究发现,17OHD患者体内皮质酮水平可高达正常值的50~100倍,而DOC水平可达正常人的1 000倍[11]。高水平皮质酮可弥补皮质醇的不足,因此17OHD患者往往不会出现糖皮质激素不足,甚至肾上腺危象,疾病早期难以被识别,临床上易漏诊[12]。而去氢表雄酮、睾酮及雌激素等合成障碍会出现性发育异常。关于CYP17A1的基因型和表型之间的关系尚不清楚,截至目前超过100种CYP17A1基因突变被报道,包括点突变、小的插入或缺失、剪切位点的改变及少数大范围的缺失[13]。研究发现,R96W、R347H/C和F417C是西方国家较为常见的突变类型,W406R和R362C突变在巴西地区流行更为广泛,p.H373L突变在亚洲人群中更为常见[14]。17OHD是我国第二常见的CAH,我国突变类型以缺失突变+替代突变的复合类型为主,其中p.Y329fs和D487_F489是我国最常见的突变类型,超过80%的患者都是因这两种突变所引起的。研究也发现,p.Y329fs、D487_F489具有“祖先效应”,即携带致病基因的杂合子祖先,由于近亲结婚使纯合子频率增高,进而使疾病发病率增加,其中p.Y329fs突变最初于2003年由Hahm等发现,随后便在中国广泛报道。国内外报道第6、8号外显子是基因突变的高发区,在临床工作中对拟诊患者可作为首选检测。
3 临床表现
17OHD患者的临床表现复杂多样,无论是46,XX还是46,XY患者均为女性表型,高血压、四肢无力通常是这种疾病的早期信号,大约90%的患者都会出现高血压和(或)低钾血症,但是仍有10%~15%的患者血压正常[15-17]。根据酶失活的程度将17OHD分为[18]:(1)完全型:完全型17OHD患者因17α-羟化酶和17,20裂解酶活性完全丧失,皮质醇和性激素合成受阻,ACTH分泌代偿性升高导致DOC等有盐皮质激素作用的前体物质累积,因此患者常会出现高血压、反复低血钾。此外,性类固醇可诱导骨骺闭合延迟,因此骨龄常常落后于正常同龄人;促性腺激素的长期升高而雌激素缺乏,发生卵巢囊肿和囊肿破裂等。一般无明显糖皮质激素缺乏症状,罕见肾上腺危象发生。染色体为46,XX的患者特点为原发性闭经、第二性征缺如、性器官幼稚,其他还可包括卵巢囊肿和囊肿破裂;染色体为46,XY的患者特点为异位(腹腔或腹股沟等)发育不良的睾丸、外观呈女性幼稚型或假两性畸形、阴道盲端。(2)部分型:极少数17OHD患者因17α-羟化酶/17,20裂解酶活性部分丧失,因此保留了部分雌激素和雄激素功能,高血压、低钾血症的程度较完全型轻;46,XX患者可出现不同程度的第二性征发育,46,XY的患者可有轻度阳痿。
4 诊断及鉴别诊断
17OHD因早期症状不典型,且发病率低,早期诊断具有一定困难,据统计约88%的病例直到青春期甚至更晚才得到确诊[3]。诊断主要依据特征性的临床表现、类固醇成分分析及ACTH兴奋试验。具体参照中华医学会第十一次全国内分泌学学术会议确定的诊断标准(2012年8月)[19]:(1)完全型17α-羟化酶/17,20裂解酶联合缺陷症:患者为女性表型且伴有第二性征缺乏、原发性闭经、高血压、低钾血症等表现,皮质醇水平低于参考值,ACTH水平反馈性增高;性激素(雌二醇、睾酮)明显低于参考值而促性腺激素(LH、FSH)水平升高可诊断。(2)部分型17α-羟化酶/17,20裂解酶联合缺陷症:根据患者外生殖器存在假两性畸形或有自发的青春期第二性征发育及月经,或血压和血钾水平正常,结合体内可检测出17α-羟化酶,并且对ACTH兴奋试验有反应可予以诊断。对于所有高血压、低钾血症并伴有性发育异常同时影像学发现肾上腺皮质增生,X线提示骨龄落后、骨质疏松的患者要引起临床医生的高度重视,怀疑17OHD,必要时行基因检测。同时需与原发性醛固酮增多症、Turner综合征、P450-氧化还原酶缺乏症(PORD)以及17β-羟类固醇脱氢酶3型(17β-HSD3)缺陷症等鉴别[20]。原发性醛固酮增多症患者的典型表现为高血压伴低血钾,影像学CT检查常表现为单侧肾上腺腺瘤,无性腺发育异常。Turner综合征患者常身材矮小、特殊体征,且伴有淋巴水肿、心脏畸形等表现,根据患者染色体核型可予以鉴别。PORD可导致以合并CYP21A2和CYP17A1缺陷为特征的PORD,是CAH的一种罕见亚型。PORD以出生时两性畸形、性激素合成障碍和特征性骨骼畸形为独特表现,临床表现多样。46,XX胎儿可出现阴蒂肥大等畸形,青春期因性激素合成障碍造成生殖器不发育或发育不良,46,XY患儿出生时由于睾酮不足,表现为小阴茎或尿道下裂,部分患儿存在隐睾,除了激素分泌紊乱,多数PORD患者伴有不同程度骨骼畸形,主要包括颅面部畸形,可与17OHD相鉴别[21]。其中PORD新生儿常伴有与之相关的骨骼缺陷,称为Antley-Bixler综合征[22]。17β-HSD3缺陷症是由于17β-HSD3缺乏或不足所致的罕见常染色体隐性遗传疾病,17β-HSD3可在睾丸组织将雄烯二酮催化成睾酮,该酶的缺陷会导致睾酮水平低下,46,XX个体表型基本正常,46,XY患者常表现为不同程度男性化不全,假两性畸形或完全为女性表型,可伴有阴蒂肥大、阴囊融合或阴道盲端,实验室检查雄烯二酮水平升高和低水平睾酮,绒毛膜促性腺激素兴奋试验有助于诊断[23]。
5 治疗和预后
激素替代是17OHD最主要的治疗方案,由于治疗周期较长,治疗过程中应严密监测患者激素水平、离子水平及生长发育情况。糖皮质激素可抑制ACTH和促肾上腺皮质激素释放激素,进而抑制盐皮质激素的分泌,使患者血压降低,血钾升高。糖皮质激素以小剂量为主,目前常用的药物包括地塞米松、强的松和氢化可的松。研究发现,0.5~1.5 mg的地塞米松可以纠正高血压、低钾血症等盐皮质激素相关症状[24]。针对儿童和青少年患者常选择氢化可的松[1]。成年患者一般选择中、长效糖皮质激素,但是长期的激素替代可能会发生骨质疏松等不良反应,可根据患者骨代谢指标及骨密度水平补充钙剂。大多数患者规律使用糖皮质激素治疗后,血压、血钾恢复正常,部分患者单用糖皮质激素血压控制不佳,需加用盐皮质激素受体拮抗剂等辅助治疗,以阻断盐皮质激素对心、肾等靶器官的毒性反应,减少靶器官的损害[25]。50~200 mg/d的螺内酯1~2次给药为最理想的治疗方案,但男性患者会出现勃起功能障碍和乳房发育等不良反应,选择性盐皮质激素受体拮抗剂依普利酮因不良反应少,可作为男性患者的治疗选择。研究显示,每天1~2次10 mg氢化可的松联合50 mg螺内酯治疗,血压和血钾水平可恢复正常且不会导致肾上腺轴的抑制和肾上腺危象的发生,钙通道阻滞剂如2.5~10 mg氨氯地平可辅助控制血压,但其存在外周水肿的不良反应。然而,对于治疗晚、依从性差或未规律治疗的患者,可能会出现长期血压控制差导致的一系列心、肾、眼等器官的并发症,或脑出血、脑梗死以及低钾血症诱发心律失常甚至危及生命,严重影响患者预后及生活质量。
近年来有研究提出,使用泵持续皮下注射氢化可的松(CSHI)可降低药物剂量及其不良反应。与口服激素对比,CSHI是一种安全且耐受良好的皮质醇替代疗法,可有效地模拟传统疗法控制不佳的17OHD患者生理性皮质醇分泌,目前关于CSHI疗法的长期效益尚需更多证据支持[26]。此外,由于确诊为17OHD的患者大多处在青春期并存在性发育异常,应根据患者的外阴特点、年龄、自我性别认知度、心理及家庭社会等因素给予个体化雌激素替代治疗、雄激素补充及外科手术干预,同时予以及时有效的心理疏导,帮助患者重建信心,以达到心理和生理的康复。46,XX患者几乎无生育可能,可用雌孕激素替代疗法诱导人工月经周期,必要时可考虑辅助受孕。46,XY患者常于12~13岁予以低剂量雄激素补充,常见如庚酸睾酮或丙酸睾酮,促进其第二性征的发育及骨骼成熟。此外,46,XY患者应常规给予手术干预切除双侧睾丸以防恶变。由于17OHD是一种单基因遗传病,干细胞和组织工程技术也可能为该疾病的治疗提供新的突破[27]。
综上所述,17OHD是由CYP17A1基因突变所致的CAH,患者常常以高血压、低钾血症、性发育异常为临床特征,因临床罕见且表现复杂,漏诊率和误诊率高。临床上对于出现高血压、低钾血症及第二性征未发育的患者,应积极进行激素测定、染色体核型分析等检查,必要时行基因测序以帮助诊断,对于确诊为17OHD的患者应及时予以激素替代治疗。临床医师应不断提高对该病的认知,早期诊断,及时治疗,加强多学科协作,从生理和心理多方面综合干预,提高患者生活质量,改善预后。
猜你喜欢
文萃报·周二版(2024年11期)2024-04-05 16:04:31
保健与生活(2023年1期)2023-05-30 10:48:04
中华实用诊断与治疗杂志(2022年2期)2022-09-02 01:47:02
热带作物学报(2020年9期)2020-10-29 07:35:39
中国现代医生(2020年12期)2020-07-04 03:03:59
广东医科大学学报(2020年6期)2020-02-06 06:00:58
家庭医学(下半月)(2019年9期)2019-10-12 08:03:58
心肺血管病杂志(2018年11期)2018-12-18 01:51:40
医药前沿(2018年25期)2018-01-17 19:48:36
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:16:42
