
碳酸铯催化苯五甲酸与CO2羧基化反应的研究
2020-11-19 02:28孙玉琴王玉高牛艳霞盛清涛
太原理工大学学报 2020年6期
孙玉琴,王玉高,申 峻,牛艳霞,刘 刚,盛清涛
(太原理工大学 化学化工学院,太原 030024)
CO2的化学利用正逐渐引起研究者的兴趣[1-3]。CO2主要通过构建新的C—C、C—N、C—O和C—H键来形成高附加值的化学品被加以利用[4]。在众多利用途径中,构建C—C键即应用CO2直接转换成羧基的反应毋庸置疑是具有经济意义和工业潜力的[5-6]。芳酸是重要的精细化学品,近年来许多研究者致力于将廉价易得的CO2直接引入芳环生成羧基,进而开创出反应流程短且绿色高效的芳酸制备方法[7]。
目前羧基化研究中底物大多是芳杂环和芳卤化合物等含有较强酸性C—H的物质[3,8],对此类反应的催化剂研究多集中在过渡金属催化剂上[7,9],但此类催化剂往往需要复杂的配体,价格昂贵且较难制备。碳酸盐由于廉价易得,且碱性和稳定性较强[10],逐渐被用于催化CO2羧基化反应[11-14]。KUDO et al[15]初步探究了苯甲酸铯在碳酸铯和甲酸铯的作用下与CO2的羧基化反应,发现产物为苯二甲酸铯和苯三甲酸铯,但是反应条件较为苛刻(380 ℃,CO2压力40 MPa)。2016年,BANERJEE et al[16]发现苯甲酸铯与CO2在碳酸铯催化下在相对温和的条件下(320 ℃,CO2压力0.8 MPa)就可发生羧基化反应,有利于此类反应的推广。
苯六甲酸是重要的精细化学品,其衍生物可作为原料合成侧链型聚酰亚胺,用在航天等高科技领域。目前苯六甲酸非常难制备,若能由相对易得的低羧基数苯羧酸(如苯甲酸或苯二甲酸等)与CO2发生羧基化反应来制备苯六甲酸,则既可实现CO2的有效利用,又可降低苯六甲酸的生成成本。由于低羧基数的苯甲酸和苯二甲酸等在羧基化反应过程中产物类型较多(可能生成苯三甲酸、苯四甲酸、苯五甲酸等),情况较为复杂,为了简化研究,本文以苯五甲酸作为研究对象,通过分子模拟和实验操作,探究苯羧酸与CO2在碳酸铯催化下发生羧基化反应的基本规律,为由低羧基数苯羧酸与CO2羧基化生成苯六甲酸的合成方法提供前期基础数据。
1 实验部分
1.1 仪器与试剂
本研究所用主要仪器:高效液相色谱分析仪(HPLC,大连依利特仪器分析公司,P1201)、液相色谱柱(美国Waters公司,X-Bridge)、磁力搅拌反应釜(湖南华思仪器有限责任公司)。所用试剂包括苯六甲酸(AR,>98.0%)、苯五甲酸(AR,>98.0%)、碳酸铯(AR,>99%)、CO2(体积分数大于99.4%)等。
1.2 实验方法
1.2.1理论计算方法
量化计算软件采用Gaussian公司的Gaussian 09程序包,通过密度泛函理论的B3LYP[17]方法对物质进行结构优化,由于体系中有后周期元素铯,故基组采用贋势基组lanl2dz[18]。在优化得到稳定构型后,在该结构的基础上进行振动频率分析及电荷布局分析,进而计算标准的热力学函数[19]。计算过程中所有收敛精度均取程序默认标准(优化分子构型的Maximum Force、RMS Force、Maximm Displacement、RMS Displacement分别小于各自默认值)。
不同温度下反应的焓变(ΔH(T))等于生成物总热焓减去反应物总热焓,见式(1);不同温度下反应的吉布斯自由能变(ΔG(T))等于生成物总吉布斯自由能减去反应物总吉布斯自由能,即式(2):
ΔH(T)=∑Hprod(T)-∑Hreact(T)=∑(E0+
Hcorr(T))prod-∑(E0+Hcorr(T))react.
(1)
ΔG(T)=∑Gprod(T)-∑Greact(T)=∑(E0+
Gcorr(T))prod-∑(E0+Gcorr(T))react.
(2)
式中:E0表示总内能;Hcorr和Gcorr分别为修正的热焓和吉布斯自由能;下角标prod和react分别表示生成物和反应物。根据热力学基本方程dG=Vdp-SdT,可得:dΔG=ΔVdp-ΔSdT。假设CO2为理想气体,则恒定温度下的吉布斯自由能变可近似为:dΔG≈ΔVdp≈ΔVgdp=-(RT/p)dp(由于气体体积的变化远远大于固体,故ΔVg近似于ΔV).积分后得到式(3),据此可以获得特定温度下压力对反应吉布斯自由能变的影响。
ΔG=ΔGθ-RTln(p/pθ) .
(3)
根据ΔG(T)=-RTlnK,可以得到反应平衡常数K与温度的关系,见式(4):
K(T)=exp[-ΔG(T)/RT] .
(4)
1.2.2实验操作
称取1 mmol的苯五甲酸与碳酸铯混合物(摩尔比1∶2.5,提前在研钵中混合均匀)或苯五甲酸铯盐(提前成盐干燥),加入0.55 mmol的碳酸铯快速研磨,将混合物置于200 mL不锈钢反应釜中。检查装置(图1)的气密性,通入CO2置换釜内空气三次,然后充入适量CO2达到预设压力,在目标条件下进行反应。当反应釜升温到设定温度时开始计时,待反应结束后,取出反应釜立刻放入冷水中。待反应釜的温度降至室温,打开放气阀,用氢氧化钠溶液吸收多余的CO2气体。将反应混合物用去离子水溶解,然后用浓盐酸酸化水溶液至pH约为2(得到相应的苯羧酸),用0.45 μm的无机滤膜进行减压抽滤除去水不溶物,定容至1 000 mL进行产物检测分析。
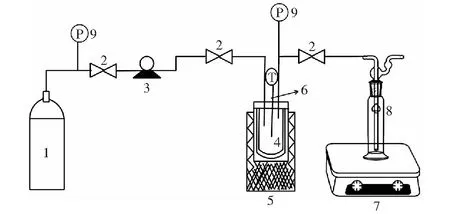
1.CO2 cylinder;2.cut-off valve;3.CO2 pump;4.reactor;5.oven;6.thermometer;7.iron stand;8.gas absorption bottle;9.pressure gauge图1 实验装置图Fig.1 Experimental device diagram
1.2.3产物分析
处理后产物用HPLC进行分析,在外标法的基础上进行定量,检测条件如下[20]:柱温为35 ℃;紫外检测波长为235 nm;流动相是体积分数0.10 %的磷酸水溶液与乙腈,流量为1 mL/min。采用二元梯度洗脱程序检测(磷酸与乙腈的起始体积比为95∶5,在10 min内降到80∶20,保持2 min后在2 min内增到95∶5).苯五甲酸与苯六甲酸标准物的液相色谱图及相应的线性关系见图2。
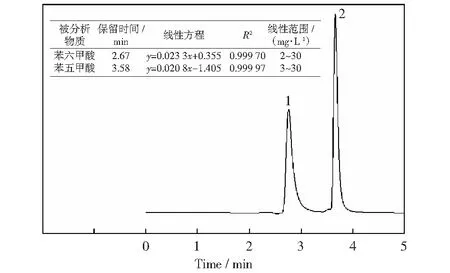
图2 苯五甲酸与苯六甲酸标准物的液相色谱图及相应的线性关系Fig.2 Liquid chromatogram and corresponding linear relationship for phenylpentacarboxylic acid and mellitic acid
2 结果与讨论
2.1 几何结构与基本性质
如图3所示,苯五甲酸羧基化生成苯六甲酸是在C2位点上进行的,故对苯五甲酸成盐前后该位点附近的结构及性质变化进行研究,得出以下结论:

图3 Gaussian程序优化的分子构型图Fig.3 Molecular configuration map optimized by Gaussian program
1) 通过计算发现苯五甲酸成盐后C2位点的空间位阻减小,这将利于反应的进行。苯五甲酸(结构a)和苯五甲酸铯盐(结构b)中的二面角数据见表1,结构a和结构b中的∠H10-C2-C3-C7及∠H10-C2-C1-C11在数值上无太大变化,说明Cs原子取代H原子后并不会使C2-H10发生明显形变。但是成盐后的结构b中∠C2-C3-C7-O8和∠C2-C1-C11-O12发生很大变化,远远大于结构a的相应二面角,这说明成盐后C1和C3位上的羧基相对于苯环平面发生了较大扭转,可能原因是Cs与邻近O之间有螯合作用,这将使C2位点空间位阻减小,有利于在C2处生成一个羧基,故苯五甲酸铯盐较苯五甲酸更容易进行羧基化反应。
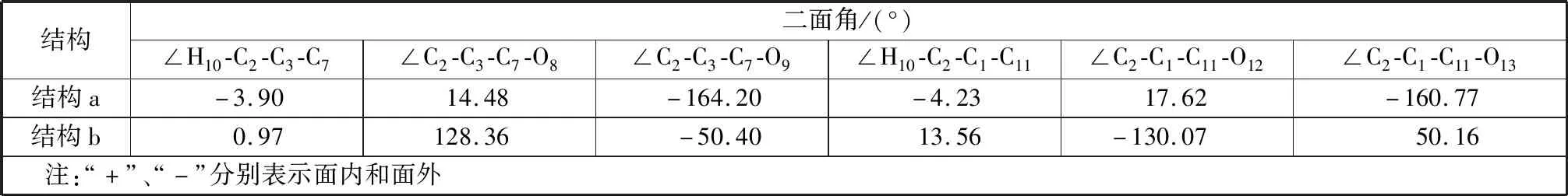
表1 Gaussian程序优化得到的苯五甲酸及其相应铯盐的二面角Table 1 Dihedral angle of phenylpentacarboxylic acid and their corresponding cesium salts optimized by Gaussian program
2) 计算结果显示苯五甲酸铯C2位电负性比苯五甲酸C2位电负性强。一般认为[18]CO2参与的羧基化反应是CO2的亲电过程,故物质的电荷布居有可能成为影响羧基化反应活性的因素。由表2可知,CO2带一定数量正电荷,C2(苯五甲酸)与C2(苯五甲酸铯)的Mulliken电荷分别为-0.474、-0.629,由此可见苯五甲酸铯反应位C的负电荷较大,有利于CO2的亲电加成。故相对于苯五甲酸,苯五甲酸铯更容易发生羧基化反应。
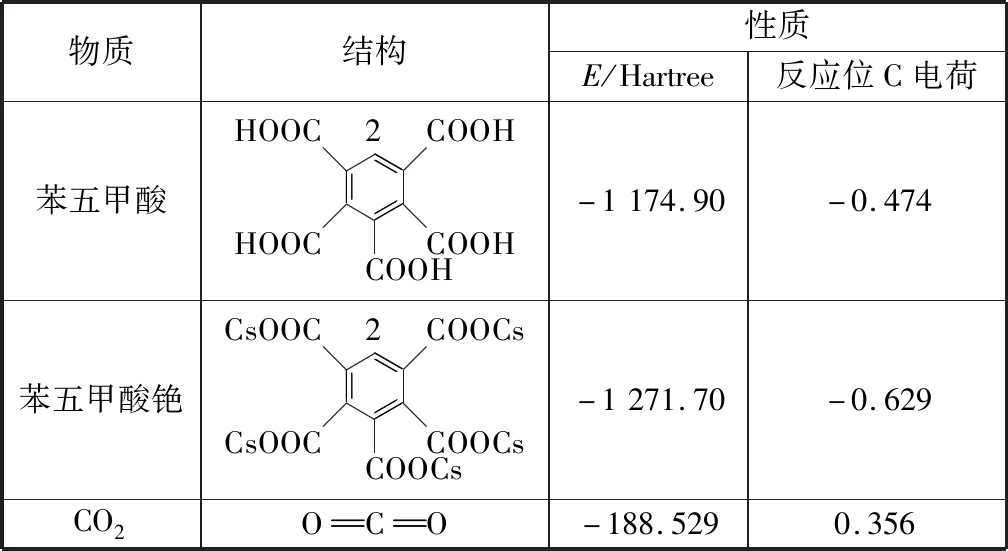
表2 苯五甲酸、苯五甲酸铯以及CO2的总分子能量及反应位C的Mulliken电荷Table 2 Total molecular energy of phenylpentacarboxylic acid, cesium phenylpentacarboxylate and CO2 and Mulliken charge of reaction site C
2.2 热力学性质
苯五甲酸铯在碳酸铯催化体系中与CO2反应生成苯六甲酸铯的反应方程式见式(5),根据式(5)计算了2.03 MPa,200~600 K温度范围内该反应的热力学性质,具体结果见表3.表中数据包括E0,Hcorr,Gcorr和Stot(熵值),四者的关系是:H=E0+Hcorr,Gcorr=Hcorr-TStot.
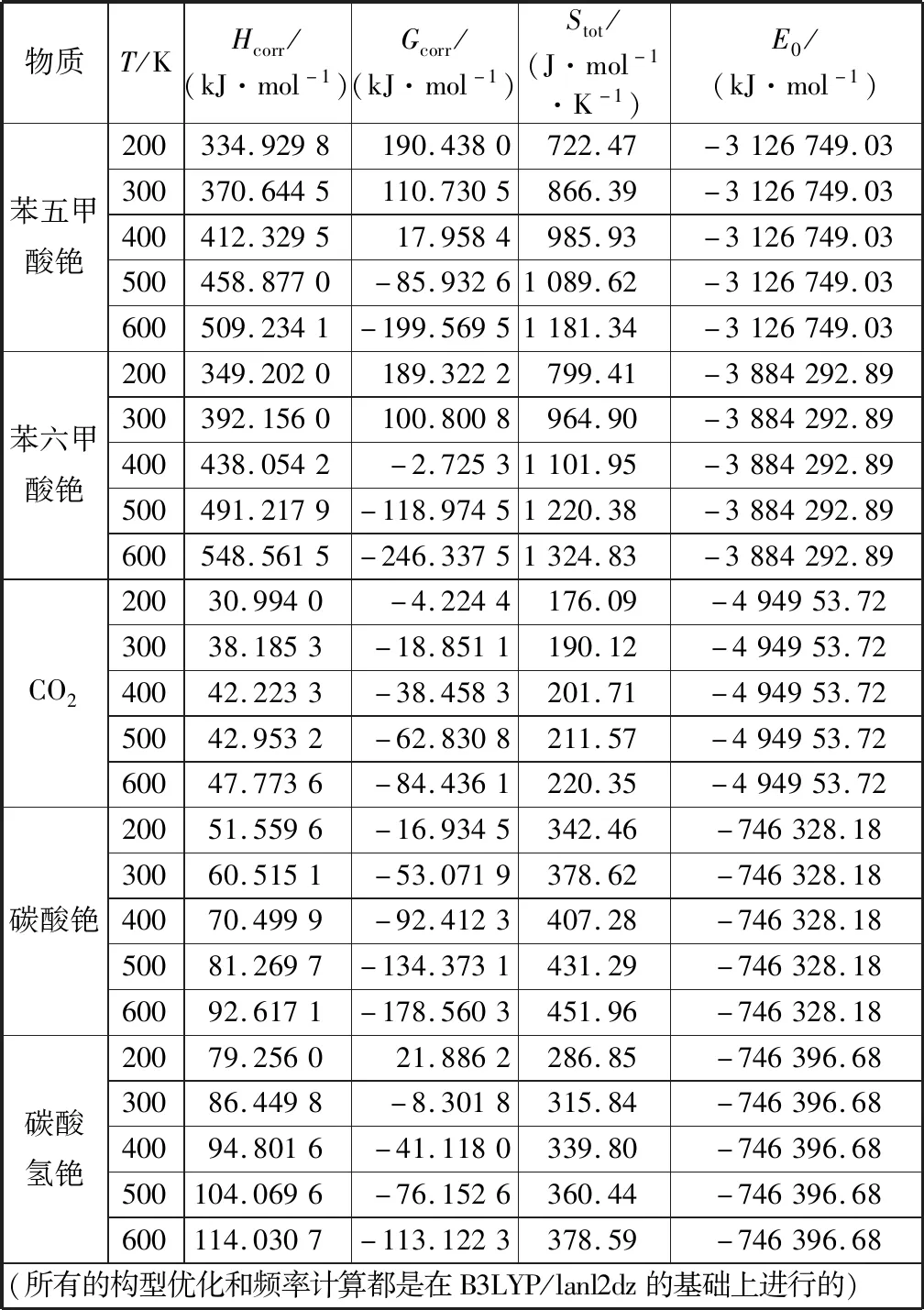
表3 反应物与产物的矫正焓、矫正吉布斯自由能、熵以及总能量Table 3 Corrected reaction enthalpy, corrected Gibbs free energy, entropy, and total energy of reactants and products
(5)
2.2.1反应的焓变及吉布斯自由能变
根据公式(1)计算反应方程(5)的ΔH,结果如图4所示。在200~600 K的范围内,反应的ΔH(T)<0,大约处于-95~-101 kJ/mol范围内,表明该反应是放热反应,且放热量较大;低温有利于反应平衡向正方向移动,但是,低温又会降低反应速度。因此,在实际反应操作中,需要综合考虑反应的平衡与反应速率两个因素,对温度进行折中选择。
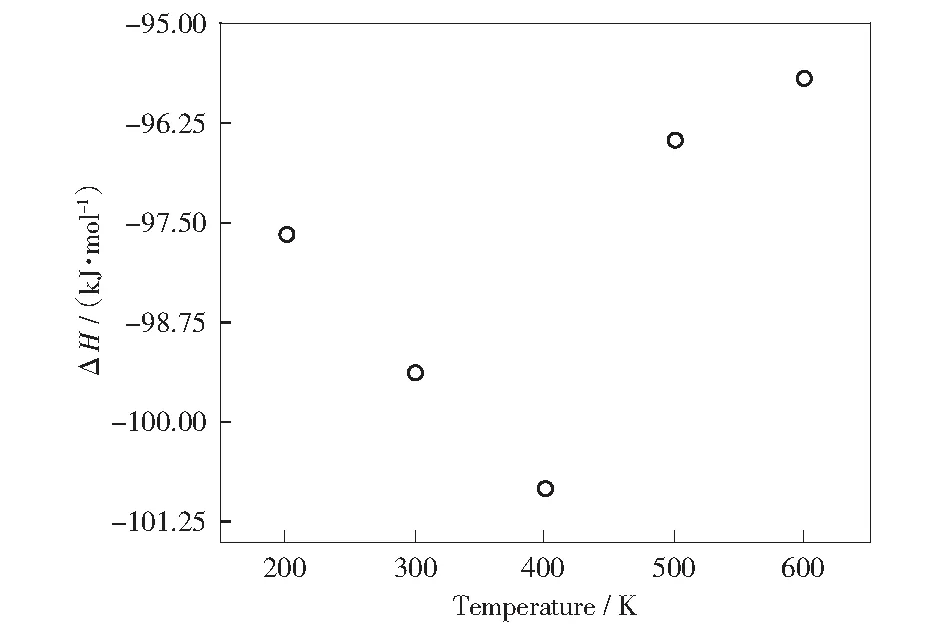
图4 苯五甲酸铯羧基化反应生成苯六甲酸铯的ΔHFig.4 ΔH of carboxylation from cesium phenylpentacarboxylate to cesium hexacarboxylate
同样,根据公式(2)计算了该反应的ΔG随温度的变化情况,结果如图5(a)所示。反应在200~600 K的范围内,ΔG(T)<0,表明反应是可以自发进行的;随着温度的升高,ΔG虽逐渐变大但仍小于0,这说明600 K仍然是反应温度的可选项,但已接近温度选择的上限。此外,由于压力对气体分子数发生改变的反应有较大影响,故需根据公式(3)进一步计算600 K时不同反应压力下的ΔG(见图5(b))。热力学表明600 K条件下,苯五甲酸铯与CO2直接羧基化生成苯六甲酸铯在压力≥2 MPa时是可自发进行的,而在1 MPa时ΔG>0,反应不可自发进行。
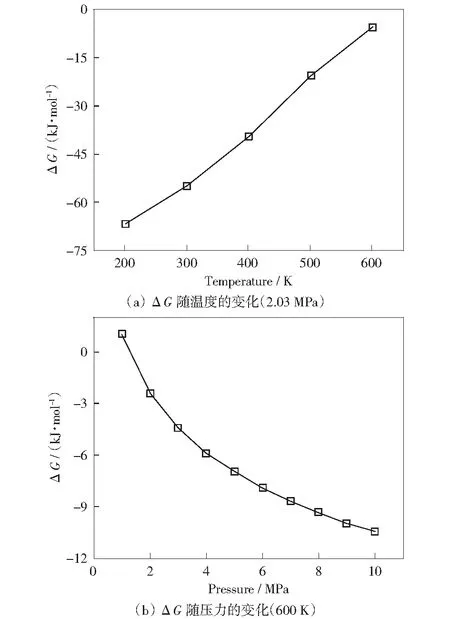
图5 苯五甲酸铯羧基化反应生成苯六甲酸铯的ΔGFig.5 ΔG of carboxylation from cesium phenylpentacarboxylate to cesium hexacarboxylate
2.2.2反应平衡常数
根据公式(4)可计算出200~600 K的温度范围内反应的平衡常数,如图6所示。反应的平衡常数随温度的升高而降低,且变化幅度较大,这说明随着反应温度的升高,羧基化反应的进行程度降低。在300 K时,lnK=22.50,反应进行程度较大;而在600 K时,lnK=1.13,K=3.10。综上所述,温度升高,不利于反应的进行。随着反应温度的升高,苯五甲酸与CO2羧基化反应进行的程度降低。
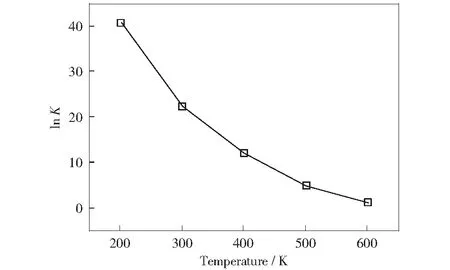
图6 苯五甲酸铯羧基化反应生成苯六甲酸铯的lnKFig.6 lnK of carboxylation from cesium phenylpentacarboxylate to cesium hexacarboxylate
2.3 反应条件对苯五甲酸羧基化反应的影响
2.3.1混合方式对苯六甲酸收率和选择性的影响
通过实验对比了混合方式对苯六甲酸收率和选择性的影响,结果如图7所示。先成盐再进行羧基化反应比研磨后直接进行羧基化效果好,苯六甲酸的选择性和收率均有很大提高。可能原因是,与研磨相比,苯五甲酸在去离子水中与碳酸铯通过成盐方式混合后分布更为均匀,更有利于其在随后的羧基化反应中与CO2接触。此外,苯五甲酸铯盐较苯五甲酸稳定,可能在羧基化反应中不易分解,进而提高其羧基化产物的收率和选择性。由前述理论计算可知,相比于苯五甲酸,苯五甲酸铯能量较低,说明其稳定性好,而且苯五甲酸铯C2反应位的电负性更大,这可能更有利于CO2的加成(见表1).
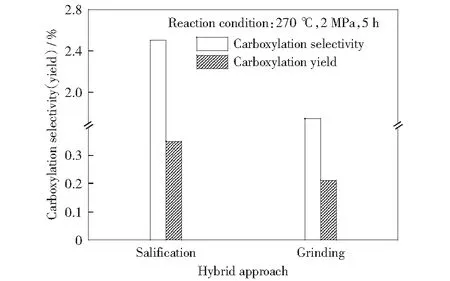
图7 成盐与研磨对苯六甲酸收率及选择性的影响Fig.7 Effect of salt formation and milling on yield and selectivity of mellitic acid
2.3.2反应温度对苯六甲酸收率和选择性的影响
通过实验对比了反应温度对苯六甲酸收率和选择性的影响,结果如图8所示。在250 ℃以下,苯五甲酸铯并不发生羧基化反应,说明苯羧酸羧基化反应需要一定的温度以使反应体系活化。随着温度的升高,羧基化产物的收率与选择性均不断提高,在270 ℃时达到最大值,但继续升高温度,收率和选择性又均下降,可能是温度太高,导致生成的苯六甲酸铯脱羧为羧基数较少的苯羧酸铯。当温度升高至320 ℃时,可能是此时的苯五甲酸铯更易脱羧,导致其转化率增大,而苯六甲酸的收率减少。故表现出来的结果是羧基化产物的收率增加,但选择性却依旧下降。
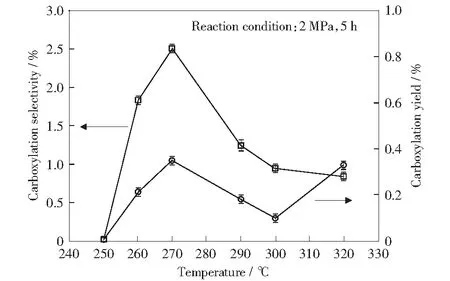
图8 温度对苯六甲酸收率及选择性的影响Fig.8 Effect of temperature on yield and selectivity of mellitic acid
2.3.3反应压力对苯六甲酸收率和选择性的影响
通过实验对比了反应压力对苯六甲酸收率和选择性的影响,结果如图9所示。随着压力的逐渐增大,苯五甲酸羧基化反应的收率与选择性先增加后降低,这说明保证充足的压力有利于反应的进行,压力的适度增加可促进体系的传质传热,且CO2作为反应原料,适当增加可促进反应的进行。但过量的压力并不利于羧基化反应,可能原因是CO2的增加减少了反应介质的极性,进而导致反应速率变慢[21];此外,CO2的增加还可使催化剂碳酸铯转变为碳酸氢铯[16],从而不利于其起催化作用,进而可能导致苯六甲酸的收率和选择性有所下降。极值点出现在4 MPa处,究其原因可能是在270 ℃时,釜内CO2达到了超临界状态,反应的传质和传热效果比较好[22],故此时羧基化反应的收率和选择性较高。
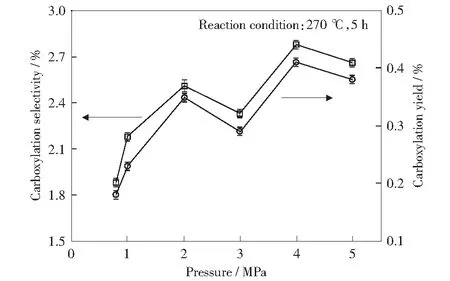
图9 压力对苯六甲酸收率及选择性的影响Fig.9 Effect of pressure on yield and selectivity of mellitic acid
2.3.4反应时间对苯六甲酸收率和选择性的影响
通过实验对比了反应时间对苯六甲酸收率和选择性的影响,结果如图10所示。随着时间的增加,苯六甲酸收率与选择性的变化趋势均是先增加后降低,这可能与CO2的扩散系数有关。在一定时间范围内,CO2可以较快速与苯五甲酸铯及碳酸铯结合,进而达到反应平衡;但反应时间进一步延长时,反应的相对速率下降[21],且反应时间较长时,生成的苯六甲酸铯可能会发生分解,故在较长时间内苯六甲酸的收率及选择性有所下降。还需注意的是收率和选择性达到极值点的时间并不一致,可能是苯五甲酸羧基数太多,副反应影响较大所致。
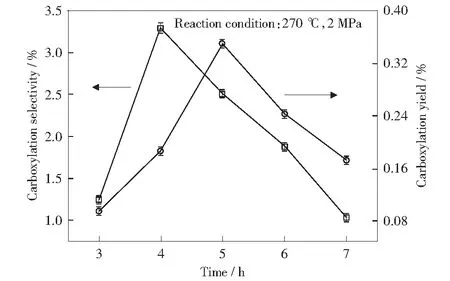
图10 时间对苯六甲酸收率及选择性的影响Fig.10 Effect of time on yield and selectivity of mellitic acid
3 结论
通过理论计算和具体实验操作探究了苯五甲酸在碳酸铯催化下与CO2合成苯六甲酸的反应,反应条件相对温和。
理论计算表明该过程在2 MPa及以上压力时是自发进行的,且该反应是放热的,平衡常数随温度的升高而降低,苯五甲酸成盐后反应位空间位阻小且负荷较多负电荷,利于其进行羧基化反应。
实验操作表明,在一定反应时间下,反应温度和压力的适度增加可使苯六甲酸的收率和选择性提高,但是时间过长,其收率和选择性又会有所下降。
后续可添加复合盐以降低体系的活化温度来提高反应活性,降低脱羧产物的收率,进而提高苯六甲酸的收率和选择性。
猜你喜欢
橡塑技术与装备(2022年8期)2022-12-17
世界农药(2022年10期)2022-11-10
中国农业科学(2022年17期)2022-09-19
云南化工(2022年2期)2022-03-18
科学与财富(2021年13期)2021-07-04
农药科学与管理(2021年2期)2021-03-16
生物工程学报(2020年6期)2020-07-31
中国科技纵横(2016年13期)2016-08-22
安徽医科大学学报(2015年8期)2015-06-01
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
