
Cu@CNT在甲醇裂解反应中构效关系的DFT研究
2020-11-19 02:28:44宫亚玲任瑞鹏
太原理工大学学报 2020年6期
宫亚玲,庞 可,任瑞鹏
(太原理工大学 煤科学与技术教育部与山西省共建重点实验室,太原 030024)
甲醇裂解反应是其高效利用不可避免的重要步骤,该反应过程中催化剂扮演了重要的角色,但是催化剂的积碳和CO中毒问题制约了甲醇高效利用。ZSM-5分子筛被认为是甲醇裂解反应的有效催化剂,但存在石墨碳覆盖活性位点和多环芳烃堵塞孔道等积碳问题,影响了分子筛催化剂的工业化应用[1-3]。为了提高分子筛催化剂在甲醇裂解过程中的催化性能,研究者采用金属对分子筛催化剂进行改性,金属改性后多环芳烃的形成明显减少[4],但是仍无法避免酸性位被覆盖。SAPO-34分子筛也是甲醇裂解的有效催化剂,目前影响其工业化应用的原因主要是积碳失活,积碳主要来源于反应过程中碳的聚集[5-6]。此外贵金属铂也是甲醇裂解中出色的单金属催化剂,甲醇在金属铂上裂解的主路径为CH3OH→CH3O→CH2O→CHO→ CO,反应过程中不会造成碳的积聚,但铂的活性催化位点容易被CO覆盖造成中毒[7-9]。综上所述,可以发现:催化剂积碳形成的原因主要为甲醇裂解过程中生成的碳聚集覆盖在催化活性位点;CO中毒的原因则为CO因其吸附能过大难以从催化剂表面解吸,沉积在表面引起中毒。因此,甲醇高效利用的前提是开发出活性中心与反应介质隔离且CO容易脱附的稳定催化剂。
研究表明,限制在碳纳米管通道内的金属纳米颗粒与沉积在碳纳米管管壁的相同金属具有不同的催化活性以及稳定性[10-14]。CUI et al[15]发现,将碳化钼纳米颗粒封装在单壁碳纳米管中用于析氢反应,封装于碳纳米管内的碳化钼比分散在碳纳米管外壁上的碳化钼纳米颗粒表现出更高的催化活性和稳定性。ZHANG et al[16]将Pt簇封装在碳纳米管通道中用于甲苯氧化反应,研究发现:碳纳米管的封装作用可以保护Pt簇免受氧气的影响;与沉积在碳纳米管外壁和炭黑载体表面的Pt簇相比,碳纳米管封装的Pt簇具有更高的活性和稳定性;碳纳米管缺乏强酸位点,避免了反应过程中的碳沉积和孔道堵塞。显然,碳纳米管封装纳米金属颗粒(M@CNT)做催化剂可以将活性中心与反应介质隔离,提高金属催化剂的活性和稳定性。基于碳纳米管封装金属的这一特性,M@CNTs催化剂可能是甲醇裂解的优选催化剂。
有关甲醇裂解的金属催化剂种类有很多,其中Cu基催化剂被认为是甲醇裂解的有效催化剂[17-19]。MEHMOOD et al[20]探索了甲醇在Cu4簇上的裂解,发现甲醇在Cu4簇上首先通过C—H键断裂生成CH2OH,随后通过C—H键断裂最终生成CO,CO的吸附能为-1.71 eV,其易从催化剂表面解吸,反应过程中无CO中毒问题。Garcia-Muelas et al[17]研究了甲醇在Cu(111)、Ru(0001)、Pt(111)和Pd(111)表面的分解,其中Cu(111)优先生成CH2O,而Pt(111)、Pd(111)和Ru(0001)最初生成大量的CO,可能会毒化表面。CO在这些金属催化剂表面的吸附能分别为-0.25 eV、-2.78 eV、-3.21 eV和-2.04 eV,可见与其他金属相比,Cu基催化剂具有优异的CO耐受性。CO在Cu表面的低吸附能是催化剂抗中毒的主要原因。
本文运用密度泛函理论研究了甲醇在Cu@CNT上的裂解机理,并探索了该催化剂的抗积碳性及CO耐受性。
1 计算模型及方法
1.1 计算模型
关于碳纳米管的选择,XIAO et al[21]发现封装金属的碳纳米管管径对催化活性的影响呈火山型曲线关系,内径约1.0 nm的碳纳米管应为最佳碳纳米管。本文构建了直径为0.93 nm的(12,0)碳纳米管,并将Cu原子封装在碳纳米管管道内。优化后的构型如图1所示,封装的Cu原子与碳纳米管管壁的C原子成键,C—Cu键长为0.208 7 nm.催化剂表面有四种不同的吸附位点,分别为与Cu原子成键的C原子的顶位(T1)、T1邻位C原子的顶位(T2)、桥位B和穴位H。

图1 Cu@CNT催化剂最优结构的侧视图和俯视图Fig.1 Side view and top view of the optimal structure of Cu@CNT catalyst
1.2 计算方法
本研究中所涉及到的所有计算均采用基于密度泛函理论的VASP软件包(Vienna abinitio simulation package)进行[22]。采用缀加投影波函数(PAW)来描述电子-离子的相互作用[23],并使用广义梯度函数(GGA)PEB计算交换相关能[24]。平面波截断能和布里渊区K点分别设置为270 eV和3×2×2.通过计算甲醇及其裂解过程中涉及到的中间体的吸附能来确定各物种的最佳吸附位点。吸附能Eads使用以下定义式计算:
Eads=ECu@CNT+adsorbate-ECu@CNT-Eadsorbate.
(1)
式中:ECu@CNT+adsorbate是催化剂Cu@CNT和吸附物的总能量,ECu@CNT是Cu@CNT催化剂的能量,Eadsorbate是吸附物的能量。通常Eads为负值时表示该吸附过程为放热吸附或稳定吸附,且Eads绝对值越大表示被吸附物与催化剂Cu@CNT表面之间的相互作用越强,Eads为正值时则相反[25]。采用NEB法搜索甲醇在Cu@CNT表面裂解过程中所有基元反应的过渡态(TS),并通过虚频计算进行识别[26-27]。活化能垒(Ea)的计算公式为:
Ea=ETS-EIS.
(2)
式中:ETS、EIS分别为各基元反应中过渡态(TS)和初始态(IS)的能量。速率k可通过传统过渡态理论计算,k值与指前因子(A)和活化能(Ea)有关[28],如式(3).R和T为气体常数和温度(本文中为298 K),kB和h是普朗克常数和玻尔兹曼常数,QIS和QTS分别为初始状态和过渡状态的分配函数。
(3)
2 结果与讨论
2.1 甲醇及其他中间体的吸附
通过研究甲醇及其他中间体在几个不同吸附位点的吸附情况,确定了各物种的最优吸附位点。吸附能和吸附构型的相关结构参数列于表1中,构型图如图2所示。

表1 Cu@CNT表面甲醇裂解过程中各物种的吸附能及结构参数Table 1 Adsorption energies and structural parameters of various species during methanol decomposition on Cu@CNT
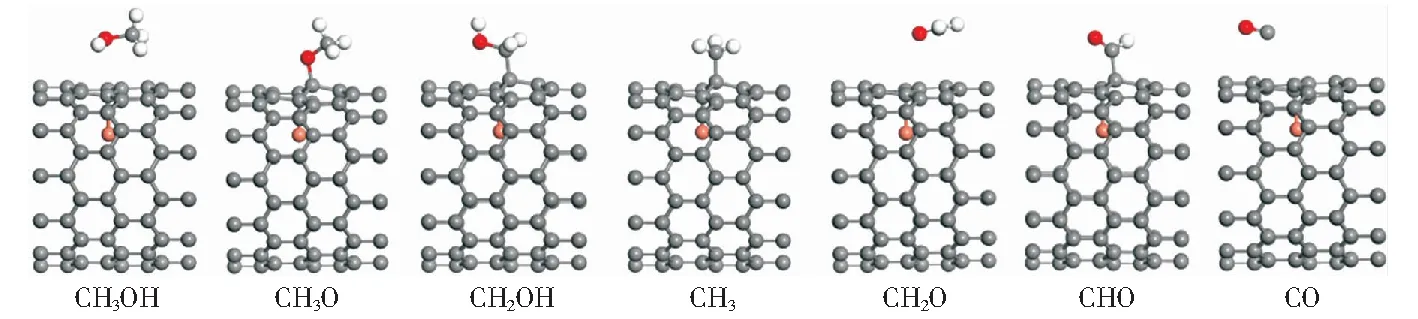
图2 Cu@CNT表面甲醇裂解过程中各物种的稳定吸附构型Fig.2 Most stable adsorption configurations of various species during methanol decomposition on Cu@CNT
2.1.1CHxOH(x=2,3)
计算结果表明,甲醇的最稳定吸附构型是通过O原子稳定吸附在Cu@CNT面的T1位点,其吸附能为-0.28 eV,甲醇在Cu(110)[29]、Cu(100)[30]和Cu(111)[31]表面的吸附能分别为-0.49 eV、-0.44 eV、-0.36 eV。此外,气相中甲醇的O—H键、C—O键和C—H键的键长分别为0.096 1 nm、0.142 5 nm和0.111 0 nm,吸附后甲醇的O—H键和C—O键伸长到0.096 6 nm和0.145 6 nm,而C—H键缩短到1.105 nm,可见吸附后甲醇变化较小,表明甲醇在Cu@CNT表面的吸附较弱。羟甲基(CH2OH)是甲醇裂解过程中的重要中间体。CH2OH更倾向于通过C原子吸附在Cu@CNT面的T2位点,吸附能为-2.74 eV,而CH2OH在Cu(110)[29]和Cu(100)[30]表面的吸附能分别为-1.97 eV和-1.83 eV.C—O键和C—C键的键长分别为1.454 nm和1.572 nm.
2.1.2CHxO(x=0,1,2,3)
如图2所示,CH3OH经O—H键断裂形成的甲氧基(CH3O)优选通过O原子吸附在Cu@CNT面T2位点,吸附能为-2.44 eV.CH3O中的O原子与Cu@CNT表面的C原子形成的C—O键与管轴方向垂直,键长为0.149 9 nm.此外,吸附物CH3O中C—O键键长(0.145 8 nm)比自由基CH3O中的C—O键(0.139 6 nm)长0.062 nm.CH2O由CH3O中C—H键断裂形成,其最稳定的吸附构型是通过O原子吸附在Cu@CNT表面的T1位点,吸附能为-0.38 eV.此外,CH2O中C—O键平行于催化剂管轴方向。CHO的最优吸附位为T2位,吸附能为-1.52 eV,CHO在Cu(110)[29]面和Cu(100)[30]面的的吸附能分别为-1.52 eV和-2.23 eV.C—O键和C—H键的长度分别为0.121 1 nm和0.112 9 nm.此外C—O—H所形成的键角为122°.CO优选以通过C原子吸附在Cu@CNT面的T2位点,吸附能为-0.47 eV,而CO在Cu(110)面[29]、Cu(100)面[30]和Cu(111)面[31]的吸附能分别为-1.21 eV、-1.04 eV和-0.98 eV.
如表1所示,CH3OH、CO及CH2O的吸附能远低于其他物种的吸附能,产生此结果的原因是自由基中间体中具有未成对电子,与Cu@CNT的相互作用强于分子中间体。此外,CO的吸附能为-0.47 eV,有助于其从催化剂表面脱附,提高催化剂对CO中毒的耐受性。Cu@CNT催化剂中C—Cu键基本稳定在0.208 7 nm左右,表明该催化剂构型是稳定的。
2.2 甲醇裂解反应
本部分讨论了甲醇在Cu@CNT催化剂作用下的裂解机理。甲醇的裂解是从其以气态形式吸附到Cu@CNT催化剂表面开始的。甲醇有三种可能的裂解路径,如图3所示:路径1,甲醇通过O—H键断裂生成CH3O中间体,最终生成CO或C;路径2,甲醇通过C—H键断裂生成CH2OH中间体,最终生成CO或C;路径3则为甲醇通过C—O键断裂生成CH3中间体,随后通过C—H键断裂产生C.各基元反应的反应热、活化能垒及速率如表2.主反应路径中各基元反应的构型图如图4所示。
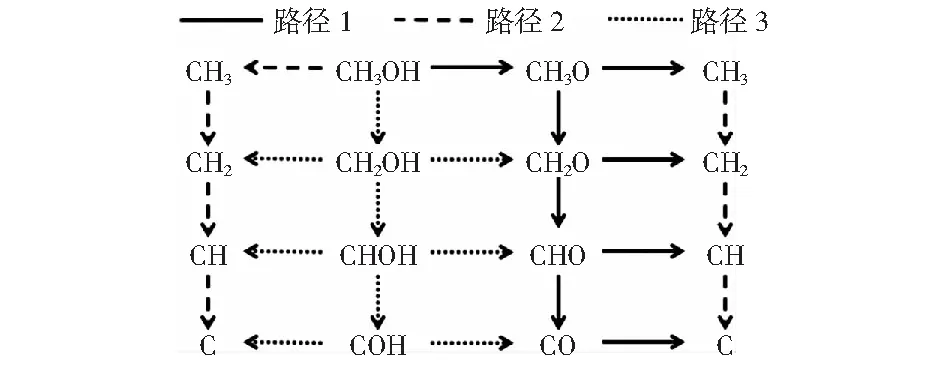
图3 Cu@CNT表面甲醇裂解的可能路径Fig.3 All possible paths of methanol decomposition on Cu@CNT
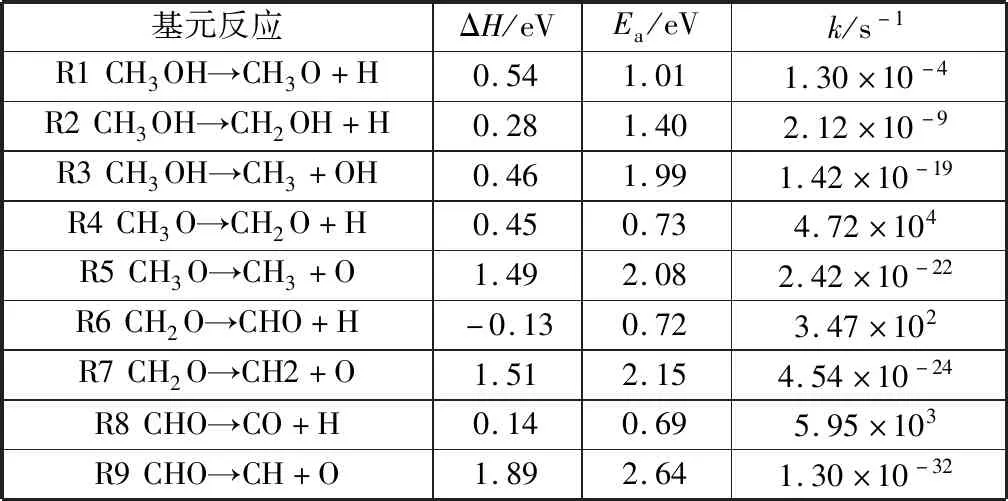
表2 Cu@CNT表面甲醇裂解过程中各基元反应的反应热ΔH、活化能垒Ea和在298 K下的速率kTable 2 Reaction energy ΔH, activation energy barrier Ea, and rate constant k at 298 K for the elementary steps involved in methanol decomposition on Cu@CNT

图4 Cu@CNT表面甲醇裂解主路径反应的IS,TS和FS的优化构型Fig.4 Optimized configuration of IS, TS, and FS for methanol decomposition reaction on Cu@CNT
2.2.1CH3OH的裂解
甲醇初始裂解有以下三种不同的路径:分别为O—H、C—H和C—O键断裂。路径1始于吸附在T1位的CH3OH,终于通过O原子吸附在T2位的CH3O和吸附于T1邻位的H.在TS1中,O—H键长从IS中的0.096 6 nm增加到0.119 7 nm.该过程的活化能为1.01 eV,反应热为0.54 eV.而在Cu(100)表面CH3OH中C—H键断裂的活化能为1.60 eV[30].在路径2中甲醇通过C—H键断裂生成CH2OH和H.通过O原子吸附于T1位的CH3OH作为IS,吸附于T2位点CH2OH和吸附于T1位的H原子作为FS.C—H键长从IS的0.110 8 nm伸长到TS中的0.164 2 nm.计算结果表明该基元反应的活化能为1.40 eV,反应热为0.28 eV.CH3OH还可通过C—O键断裂生成CH3和OH,CH3OH中C—O键被活化生成通过C原子吸附于T2位点的CH3和垂直吸附于T2对位的OH.该基元反应的活化能为1.99 eV.
甲醇初步裂解的势能图如图5所示。比较甲醇初始裂解的三种不同的路径发现,CH3OH通过O—H键断裂形成CH3O和H,比通过C—H键断裂形成CH2OH和H和通过C—O键断裂形成CH3和OH具有更低的活化能,因此甲醇初步裂解优选通过O—H键断裂生成CH3O.此外,O—H键断裂(1.30×10-4s-1)的速率远大于C—O键(1.42×10-19s-1)和C—H键(2.12×10-9s-1)的断裂速率。因此后续只讨论路径1.
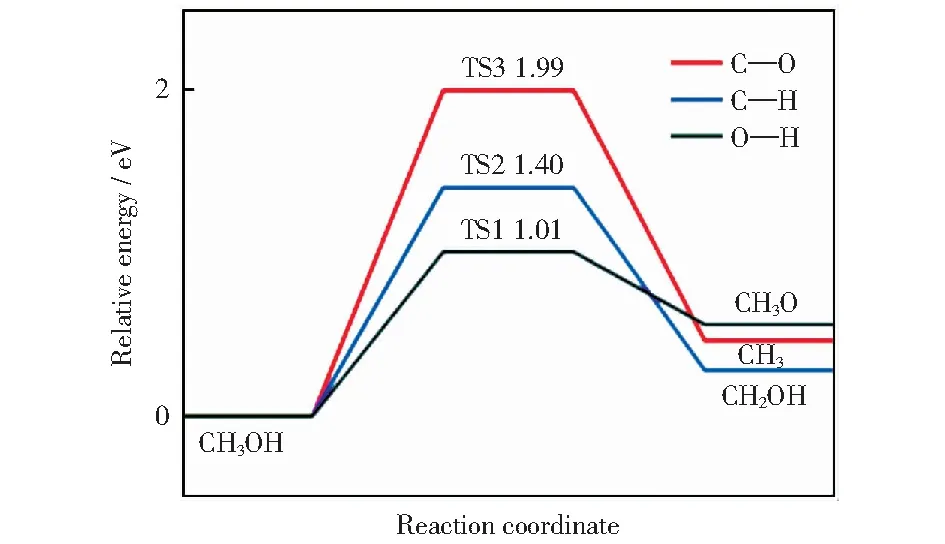
图5 Cu@CNT表面甲醇初始裂解的势能图Fig.5 Potential energy profile of methanol initial decomposition on Cu@CNT
2.2.2CH3O的裂解
CH3O的后续裂解存在两种路径。一种是CH3O脱氢形成CH2O,另一种则是通过C—O键断裂形成CH3和O,势能图如图6所示。
对于CH3O脱氢,吸附于T2的CH3O作为IS.在FS中,CH2O从表面上升,并且H原子移动至T1邻位。C—O键逐渐向Cu@CNT表面倾斜(角度α从IS的28°增大到TS的69°),使得C—H键更加靠近催化剂表面以促进其活化。该过程的反应热和活化能分别为0.45 eV和0.73 eV.而CH3O在Cu(100)表面和Cu (111)表面C—H键断裂的活化能分别为3.08 eV[30]和1.85 eV[31].对于C—O键的断裂,活化能为2.08 eV,远高于C—H断裂的活化能,表明对于CH3O难以发生C—O键的断裂。此外,在298 K时C—H键和C—O键断裂的速率分别为4.72×104s-1和2.42×10-22s-1.
2.2.3CH2O的裂解
如图6所示,在Cu@CNT表面,CH2O的不同分解路径会生成两种不同的中间体,分别为CH2和CHO.CH2O通过脱氢反应可生成CHO和H原子,在过渡态TS6中C—H键长从IS中的0.112 7 nm延长到0.128 7 nm,FS中CHO和H原子分别吸附在T2位和T1邻位。该反应过程的活化能为0.72 eV,同时反应热为-0.13 eV.而CH2O在Cu(100)表面和Cu(111)表面C—H键断裂的活化能分别为0.85 eV[30]和1.15 eV[31].使CH2O通过C—O键断裂生成CH2和O需要克服2.15 eV的活化能,同时反应热为1.51 eV.由此可知,与C—H键断裂相比,C—O键断裂不是CH2O裂解的可能路径。此外反应速率结果显示C—H键断裂的速率(3.47×102s-1)是C—O键断裂速率(4.54×10-24s-1)的1026倍。
2.2.4CHO的裂解
CHO在T2位点生成后,可进一步通过C—H断裂生成CO和H,或通过C—O键断裂生成CH和O,势能图如图6所示。对于C—H键的断裂,该过程从吸附在T2位点的CHO中C—H键的伸缩振动开始。C—H键长从IS中的0.112 9 nm伸长到TS中的0.130 0 nm,O—C—H键角从IS中的122.55°增大到TS中232.55°.在FS中,H原子吸附于T2位点,而CO移动到T3位点。该过程的活化能为0.69 eV,反应热为0.14 eV.而CHO在Cu(100)表面C—H键断裂的活化能为0.84 eV[30]。此外,C—O键断裂的活化能为2.64 eV.与CHO脱氢相比,由于C—O键断裂的活化能远高于C—H键断裂的活化能,CHO更易发生脱氢反应。CHO中C—H键和C—O键断裂的速率分别为5.95×103s-1和1.30×10-32s-1.

图6 Cu@CNT表面CH3O后续裂解的势能图Fig.6 Potential energy profile of CH3O subsequent decomposition on Cu@CNT
综上所述,甲醇在Cu@CNT上裂解的最优路径是CH3OH→CH3O→CH2O→CHO→CO,限速步骤是CH3OH→CH3O.
3 结论
本文运用密度泛函理论探索了甲醇在Cu@CNT表面的裂解反应机理,得到以下结论:甲醇稳定吸附于T1位点,其他物种则稳定吸附于T2位点。对于甲醇的初始裂解,其更易通过O—H键断裂生成CH3O,其后CH3O逐步脱氢最终生成CO,其中O—H键的断裂是限速步骤。且在Cu@CNT表面,甲醇裂解主反应路径中各基元反应的活化能分别为1.01 eV、0.73 eV、0.72 eV和0.69 eV.甲醇在Cu(100)表面裂解的主路径与甲醇在Cu@CNT表面裂解的主路径相同,各基元反应的活化能分别为1.60 eV、3.08 eV、0.85 eV和0.84 eV,故可知Cu@CNT催化剂的催化活性高于Cu(100)催化剂的催化活性。此外,甲醇裂解的最终产物CO在Cu@CNT和Cu(100)表面的吸附能分别为-0.47 eV和-1.04 eV,故Cu@CNT具有更高的CO耐受性。不同于活性位点裸露于反应介质中的Cu(100)催化剂,Cu@CNT催化剂中碳纳米管的封装作用使活性中心和反应介质隔离,反应过程中无积碳的生成。由此可知,碳纳米管封装金属粒子有效地增强了金属催化剂的活性和稳定性,是甲醇裂解的高效稳定催化剂。
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
科学导报(2018年30期)2018-05-14 12:06:01
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:52
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
分析测试学报(2015年8期)2016-01-13 06:19:35
中学化学(2015年8期)2015-12-29 07:32:44
原子与分子物理学报(2015年3期)2015-11-24 12:49:39
Chinese Journal of Chemical Engineering(2014年3期)2014-07-24 15:40:13
火炸药学报(2012年4期)2012-01-29 07:32:58
