
超高效液相色谱串联质谱法同时测定减肥类保健食品中10种非法添加利尿类及泻下类药物
2020-11-18 03:47陈玉龙饶雅琨董曼曼王蓓蓓贾寒冰梁红月张亚锋西安市食品药品检验所陕西西安70054陕西省药品技术审查查验中心陕西西安70065
食品工业科技 2020年22期
李 卓,陈玉龙,孙 晓,饶雅琨,董曼曼,王蓓蓓,贾寒冰,王 涛,梁红月,张亚锋(.西安市食品药品检验所,陕西西安 70054;.陕西省药品技术审查查验中心,陕西西安 70065)
随着人们生活水平的提高,对美丽、健康等方面有了更多的要求,众多减肥类保健食品产品应运而生。然而许多不法分子在这些利益驱使下,追求短暂减肥效果和高额利润,在保健品中非法添加利尿类和下泻类药物,这些药物作为处方药有严格的服用剂量要求,患者在不知情的情况下长期服用这些产品时,可能会因为摄入过量引起严重的不良反应,与有些药物同服还会发生药物相互作用,导致难以预测的严重后果,使得广大消费者在身体上受到不同程度的伤害。
随着高精密仪器的发展,现已出现了许多针对违法添加药物的分析方法[1],有薄层色谱法[2]、高效液相色谱法[3-6]、超高效液相色谱-串联质谱法[7-16]、超高效液相色谱-静电场轨道肼质谱[17-18]、超高效液相色谱-四级杆飞行时间质谱法[19-21]等。显而易见,液质已成为了违法添加药物检测的主流方法。目前国内外有关保健食品的违法添加药物测定报道较多,但尚无针对减肥类保健食品同时测定10种非法添加药物(氯噻嗪、氢氯噻嗪、氯噻酮、甲氯噻嗪、吲达帕胺、酚酞、芦荟大黄素、比沙可啶、大黄酚、大黄素)的液质方法。本实验拟采用甲醇提取,电喷雾电离源,建立多种减肥类非法添加药物的超高效液相色谱-三重四级杆质谱(UPLC-MS/MS)测定方法,以便市场监管部门在此类监测检查中能发现更多的非法添加物,为广大群众的身体健康做出更安全的保障。
1 材料与方法
1.1 材料与仪器
氯噻嗪(CAS号:58-94-6,纯度100%)、氢氯噻嗪(CAS号:58-93-5,纯度99.7%)、氯噻酮(CAS号:66258-76-2,纯度99.5%)、甲氯噻嗪(CAS号:135-07-9,纯度99.6%)、吲达帕胺(CAS号:26807-65-8,纯度97.7%)、酚酞(CAS号:77-09-8,纯度100%)、芦荟大黄素(CAS号:481-72-1,纯度98.3%)、比沙可啶(CAS号:603-50-9,纯度100%)、大黄酚(CAS号:481-74-3,纯度99.2%)、大黄素(CAS号:518-82-1,纯度98.7%) 中国食品药品检定研究院;样品采集13批减肥类保健食品 均抽自百货商店、药店;甲醇、乙腈 色谱纯,Fisher;甲酸 质谱级,aladin。
Ulimate3000-TSQ Quantiva超高液相色谱-质谱联用仪 配有Trace Finder数据处理系统,美国赛默飞科技有限公司;KQ-700VDB双频数控超声波清洗器 昆山市超声仪器公司;Milli-Q Integral 5 制水机 美国Millipore公司;ME204E 万分之一电子天平、MS105十万分之一电子天平 梅特勒-托利多仪器有限公司;台式离心机 美国Sigma。
1.2 实验方法
1.2.1 标准溶液制备 分别精密称取10种药物标准品各10 mg于10 mL量瓶中,用甲醇溶解并定容至刻度,摇匀,制成1 mg/mL单一成分标准储备液,于4 ℃避光保存3个月。
分别取上述配制好的单一标准品储备液各0.5 mL,置于50 mL容量瓶中,用甲醇定容至刻度,配制成10 μg/mL的混合标准品溶液,备用。取混合标准溶液适量,用甲醇配制系列浓度的混合标准溶液。
1.2.2 样品溶液制备 精密称取0.5 g样品(颗粒剂、片剂取适量研细;胶囊剂取内容物适量研细;口服液充分摇匀、软胶囊取适量内容物混匀;蜜饯取可食部分研细)于25 mL量瓶中,加甲醇适量,涡旋振荡,超声(频率80 kHz;功率700 W)处理30 min,放冷至室温,加甲醇定容至刻度,摇匀,6000 r/min离心5 min,取上清液1 mL,用甲醇稀释10倍,用0.22 μm有机系微孔滤膜滤过,待测。
1.2.3 色谱-质谱条件 色谱条件:Thermo Hypersil GOLD色谱柱(100 mm×2.1 mm,1.9 μm);柱温30 ℃;流速0.2 mL/min;进样体积2 μL;流动相为水(A)-乙腈(B),梯度程序洗脱为0~5 min,15% B;5~12 min,15%~60% B;12~15 min,60% B;15~17 min,60%~90% B;17~20 min,90% B;20~21 min,90%~15% B;21~25 min,15% B。
质谱条件:采用多反应检测(MRM);雾化气N2;碰撞气Ar;雾化温度300 ℃;离子传输管温度350 ℃;毛细管电压3.5 kV;鞘气流速4.57 L/min;辅助气流速11.36 L/min。各目标化合物监测离子对及相关参数设定见表1。
2 结果与分析
2.1 质谱条件的优化
采用针泵流动注射连续进样的方式进行质谱全扫描检测,分别在正、负离子模式下考察了10种目标药物的一级质谱响应,发现氯噻嗪、氢氯噻嗪、氯噻酮、甲氯噻嗪、吲达帕胺、芦荟大黄素、大黄酚、大黄素的[M-H]-峰响应强度大且稳定,酚酞、比沙可啶的[M+H]+峰响应强度大且稳定,故选择正负离子分段扫描模式进行检测。然后给予母离子不同的碰撞能量进行二级质谱扫描,对子离子进行优化,每个化合物选择2个响应值高且稳定的子离子,最终在优化条件下确定响应较强的离子作为定量离子,另一离子作为定性离子,结果详见表1。
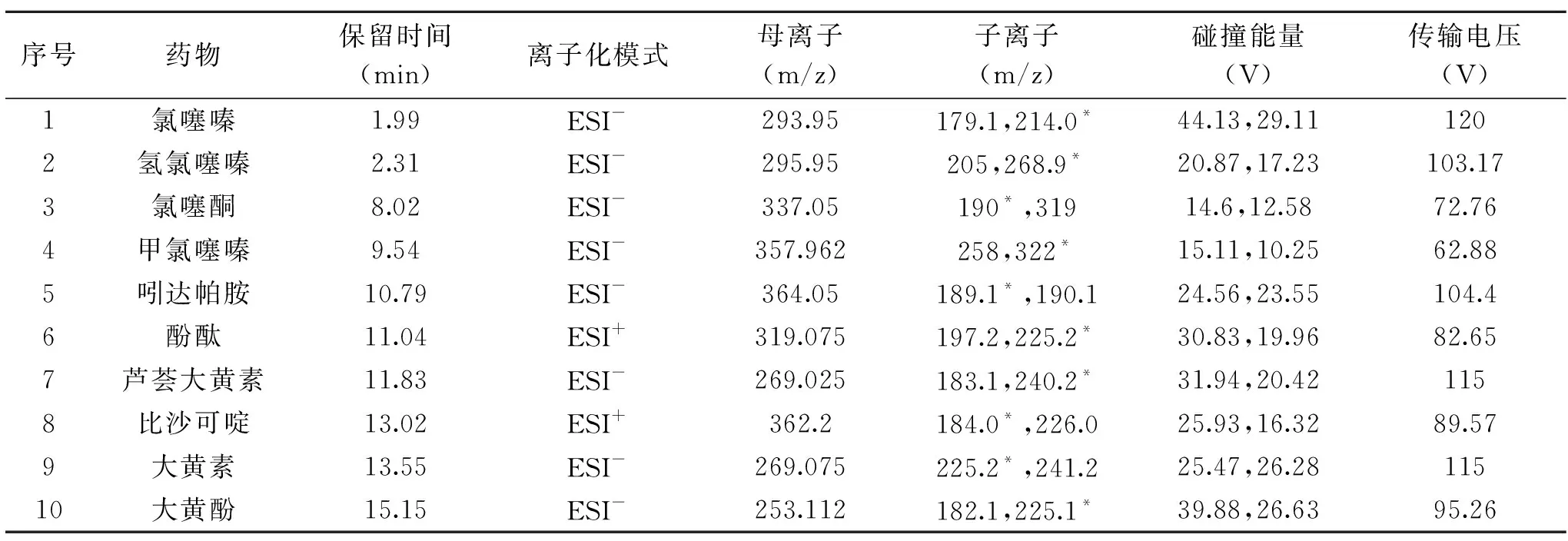
表1 10种化合物的保留时间及质谱参数Table 1 Retention time and spectrometric parameters of 10 compounds
2.2 色谱条件的优化
比较了水-甲醇、水-乙腈、0.01%甲酸水-乙腈三种体系的流动相,结果发现在水-甲醇体系中无论是以80∶20为初始流动相,还是以90∶10为初始流动相,由于溶剂效应的影响,氯噻嗪和氢氯噻嗪均为分叉峰,且都无法达到保留时间分离。在0.01%甲酸水-乙腈体系中,氯噻嗪、氢氯噻嗪、氯噻酮、甲氯噻嗪、吲达帕胺、芦荟大黄素、大黄酚、大黄素的峰响应强度有不同程度减弱。在水-乙腈体系中,10个目标化合物均有良好的峰形,且通过调节梯度洗脱程序使质荷比在3个道尔顿以内的氯噻嗪和氢氯噻嗪、芦荟大黄素和大黄素均可达到保留时间分离,从而保证了10种目标物的准确测定。提取离子流图见图1。

图1 10种化合物的多反应监测色谱图(100 ng/mL)Fig.1 Multiple reaction monitoring(MRM)chromatogram of the 10 compounds(100 ng/mL)
2.3 基质效应考察及消除
保健食品基质比较复杂,常常会干扰目标物的检测,会对检测结果的准确性产生影响。因此,在建立液质检测方法时应对基质效应进行评价,并采取消除措施,以保证结果的准确、可靠。
基质效应[22-24]是指基质成分和目标化合物在进行离子化时相互竞争而导致目标化合物信号强度有不同程度的增强或减弱的现象,包括基质增强效应和基质抑制效应。实验选用了阴性液体基质和阴性固体基质样品,各采用未用甲醇10倍稀释的空白样品溶液(BL1、BS1)、空白样品溶液(BL2、BS2)、甲醇10倍稀释空白样品溶液(BL3、BS3)加入10种药物混合标准溶液,配制成各3种混合标准基质溶液,再用甲醇(A)配制同一浓度水平的混合标准溶液,通过 MS/MS分析测定,按照式(1)计算MEx:
式(1)
式中:MEx表示基质效应的大小,大于100%为基质增强效应,小于100%为基质抑制效应。A、Bx分别为待测标物在纯溶剂和在不同样品溶液中的峰面积。
由表2可知,在BL1溶液中,氯噻嗪、氢氯噻嗪、大黄素有不同程度的基质减弱效应,其中氯噻嗪、氢氯噻嗪基质减弱较强,比沙可啶有一定程度的基质增强效应。
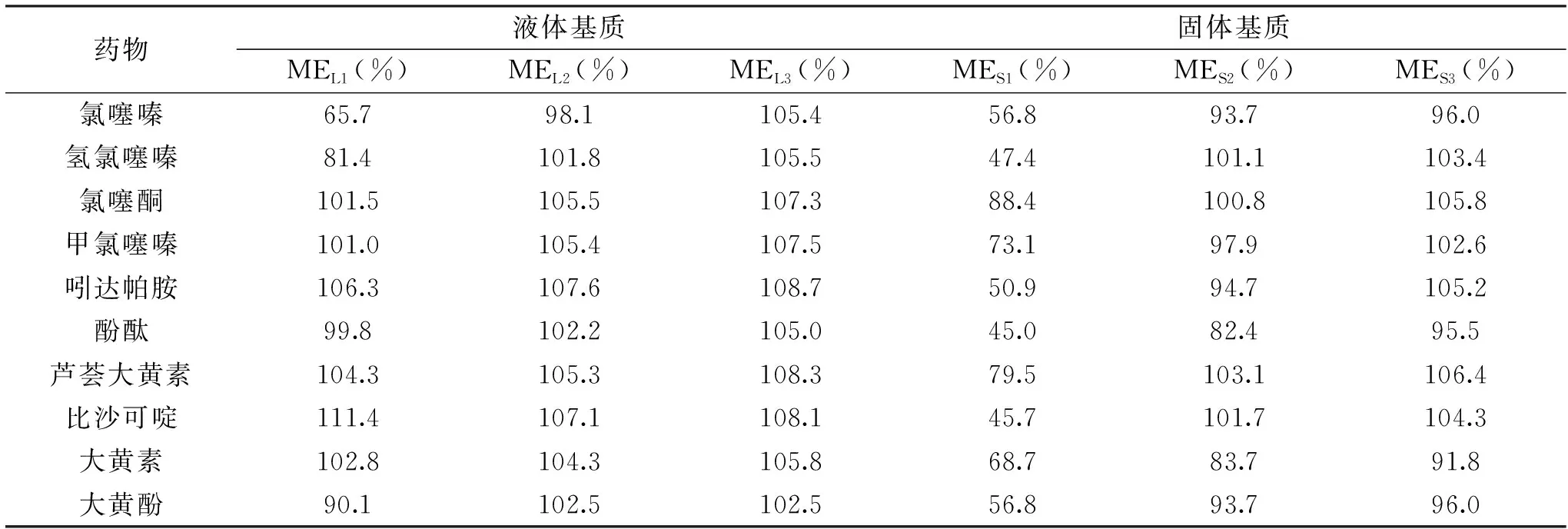
表2 10种化合物在不同稀释体积基质溶液中的基质效应Table 2 Matrix effects(ME)of the 10 compounds in different diluent
而在BL2和BL3溶液中,各化合物ME值在98.1%~108.7%之间,可忽略基质效应。在BS1溶液中,氯噻嗪、氢氯噻嗪、甲氯噻嗪、吲达帕胺、酚酞、芦荟大黄素、比沙可啶、大黄素、大黄酚均有大幅度的基质减弱效应,氯噻酮有较弱的基质减弱,可忽略基质效应。而在BS2和BS3溶液中,各化合物ME值在82.4%~106.4%之间,可忽略基质效应。故为消除基质效应带来的定量影响,最终选择样品在甲醇超声提取定容后,用甲醇十倍稀释后上机为本实验在前处理方法。
2.4 线性关系考察
取“1.2.1”项下系列浓度的混合标准溶液,按“1.2.3”项下色谱条件进行测定,以质量浓度(ng/mL)为横坐标,峰面积为纵坐标,绘制标准曲线,10种药物的回归方程及线性范围,见表3。
2.5 检测限和定量限
精密称取空白样品1份,加入混合标准溶液适量,按照“1.2.2”项下同法处理样品,按“1.2.3”项下色谱-质谱条件进行测定,以3倍信噪比(S/N)时为方法的检出下限;以10倍信噪比(S/N)时为方法的定量下限,结果见表3。

表3 10种药物的线性范围、相关系数及方法检出限、定量限Table 3 Limits of detection,limits of quantitative,linear range,regression equation and correlation coefficient of 10 drugs
2.6 仪器精密度试验
精密吸取混合标准溶液2 μL,连续进样6次,记录峰面积,计算氯噻嗪、氢氯噻嗪、氯噻酮、甲氯噻嗪、吲达帕胺、酚酞、芦荟大黄素、比沙可啶、大黄酚、大黄素的峰面积的RSD(n=6)分别为0.69%、0.57%、0.84%、2.00%、2.21%、2.98%、2.52%、1.25%、1.28%、1.79%均符合要求。
2.7 加样回收率试验
精密称取固体制剂和液体制剂空白样品各9份,加入混合标准溶液适量,并用氮气吹干,制成高、中、低三浓度三平行的加标回收样品,然后按照“1.2.2”项下同法处理该18份样品。按“1.2.3”项下检测条件进行测定,计算各组分的回收率和RSD,结果见表4。结果表明无论在固体基质还是液体基质中,10种化学成分的平均回收率均在70%~125%之间,回收率良好。

表4 10种药物回收率(n=3)Table 4 Recoveries of 10 drugs(n=3)
2.8 重复性试验
精密称取固体制剂和液体制剂阴性样品各6份(n=6),加入适量混合标准溶液,分别按“2.1.2”项下方法制备 6 份样品溶液,按“1.2.3”项下色谱-质谱条件进行测定,通过标准曲线分别计算含量,结果10中目标化合物的RSD在0.16%~8.28%,重复性较好。
2.9 稳定性试验
精密称取空白样品1份,加入混合标准溶液适量,按照“1.2.2”项下方法制备样品溶液,分别保存0、2、4、8、12、24、48 h测定10种药物的含量见图2。结果发现,氯噻酮在8 h内稳定性良好,含量变化小于4.36%,待12 h后含量变化大于10%,酚酞、甲氯噻嗪在12 h稳定性良好,含量变化小于5.93%,其余8种药物在24 h内的稳定性良好,含量变化小于6.64%。

图2 48 h内10种药物的含量Fig.2 Content of the 10 durgs within 48 h
2.10 样品测定
取13批保健食品,按照“1.2.2”项下方法制备样品溶液,分别按“1.2.3”项下色谱-质谱条件进样测定,如果样品中的质量色谱峰保留时间与标准工作液中的某种组分一致(变化范围在±2.5%之内);试样中定性离子对的相对丰度与浓度相当混合标准工作液的相对丰度一致,相对丰度偏差不超过最大允许偏差[15]规定的范围,则可判定为试样中存在该组分。结果3批检出非法添加酚酞(含量分别为534、1880、127 μg/kg)、2批检出非法添加氢氯噻嗪(含量分别为1284、462 μg/kg),阳性率为38%,其中有碧生源牌常菁茶和纤纤梅两个样品中检出少量芦荟大黄素、大黄酚、大黄素,由于产品原料中含有决明子,还需根据配料进一步计算以确定其是否为非法添加还是带入。某阳性样品的总离子流图和提取离子流图详见图3、图4。
图3 样品A的总离子流图Fig.3 Total ion chromatogram of sample A

图4 样品A的提取离子流图(酚酞)Fig.4 Extracted ion chromatogram(phenolphthalein)of sample A
3 结论
本文建立了一种减肥类保健食品中10种非法添加利尿类及泻下类药物的超高效液相-串联质谱测定方法。该方法灵敏度高,专属性好,较好地降低了基质效应带来的定量影响,适用于减肥类保健食品的非法添加药物的测定,为市场监管部门在此类监测检查中能发现更多的非法添加物提供参考。
猜你喜欢
世界最新医学信息文摘(2021年12期)2021-06-09
中华养生保健(2020年10期)2021-01-18
中华养生保健(2020年5期)2020-11-16
化工设计通讯(2020年10期)2020-09-17
海峡姐妹(2019年8期)2019-09-03
天然产物研究与开发(2019年1期)2019-03-01
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
科教导刊(2017年26期)2017-11-07
中国氯碱(2016年9期)2016-11-16
