
调整葡萄糖转运系统提高大肠杆菌L-苏氨酸产量
2020-11-11 08:55朱丽飞王小元
食品与生物技术学报 2020年8期
朱丽飞 , 王小元
(1. 食品科学与技术国家重点实验室 江南大学,江苏 无锡 214122;2. 江南大学 食品安全国际联合实验室,江苏无锡 214122;3. 江南大学 工业生物技术教育部重点实验室,江苏 无锡 214122)
关键字: 大肠杆菌;L-苏氨酸;PTS 系统;galP;ptsH;ptsG
L-苏氨酸是人体必需8 种氨基酸之一,广泛应用于食品添加剂、饲料添加剂、化妆品、医药、水产养殖和保健品等多个领域,是一种非常重要的发酵工业产品[1-3]。 大肠杆菌(Escherichia coli)作为一种模式微生物,具有遗传背景清晰、易改造、生长迅速、对发酵条件要求低等优点,最常用的是L-苏氨酸生产菌株[4-7]。
磷酸烯醇式丙酮酸(PEP)依赖的磷酸酶转移系统(PTS)是大肠杆菌中转运葡萄糖的最主要方式,通过PEP 提供磷酸,把葡萄糖转运到胞内的同时使其磷酸化成葡萄糖-6-磷酸 (G-6-P)[8-9],ptsH 基因编码PTS 系统中磷酸组氨酸搬运蛋白HPr,ptsG 基因编码膜透性酶EIICBGlc。 葡萄糖还可以通过galP基因编码的半乳糖转运蛋白GalP 进入胞内[10]。 G-6-P 由己糖磷酸异构酶PgI(pgi 基因编码)催化生成6-磷酸果糖(F-6-P),再由磷酸果糖激酶PfkA(pfkA基因编码)催化生成1,6-二磷酸果糖;由于这一步是糖酵解的关键限速步骤,pfkA 基因的表达水平一定程度上可以反映出糖酵解的强弱以及糖酵解中碳流到磷酸烯醇式丙酮酸的强弱[11-13]。 PEP 作为糖酵解的产物,由eno 基因编码的烯醇化酶催化合成,PEP 可被pykA 基因编码的丙酮酸激酶催化生成丙酮酸,丙酮酸脱氢生成乙酰辅酶A,再经gltA 基因编码的柠檬酸合酶催化生成柠檬酸,进入柠檬酸循环生成草酰乙酸。 草酰乙酸经aspC 基因编码的天冬氨酸转氨酶AspC 催化生成天冬氨酸[14],再经thrA 基因编码的天冬氨酸激酶、thrB 基因编码的高丝氨酸激酶、thrC 基因编码的苏氨酸合成酶等关键酶催化生成L-苏氨酸,见图1。
作为 PTS 系统的关键基因,ptsH 和 ptsG 的缺失或者突变会使PTS 系统不能正常转运葡萄糖,而只能利用GalP 系统等其他不消耗PEP 的方式转运葡萄糖[8,15]。 作者在一株大肠杆菌L-苏氨酸高产菌TWF001 中敲除了ptsH 和 ptsG 基因, 并且在 ptsH的敲除菌基因组上过表达galP 基因,发现改善葡萄糖转运系统有利于大肠杆菌L-苏氨酸的合成。

图1 大肠杆菌中L-苏氨酸合成代谢路程Fig. 1 Biosynthesis pathway of L-threonine in E. coli
1 材料与方法
1.1 材料与设备
本研究所有的菌株和质粒见表1, 所有的菌株都为大肠杆菌。 JM109 是用来构建质粒制作感受态用,发酵菌株TWF001 是作者所在实验室筛到的一株 L-苏氨酸高产菌, 保藏编号为 CCTCC no.M2017730[17]。 LB 培养基(g/L):蛋白胨 10,NaCl 10,酵母粉5, 固体需要添加质量分数为1.5%的琼脂。发酵种子培养基(g/L):蛋白胨 10,NaCl 10,酵母粉5,蔗糖 10,121 ℃灭菌 20 min。摇瓶发酵培养基(g/L):葡萄糖(30、40、50、60,根据需要添加),(NH4)2SO425,酵母粉 2,柠檬酸 2,KH2PO47.46,MgSO4·7H2O2,FeSO4·7H2O5,MnSO4·4H2O5,CaCO320;pH 6.8(NaOH 调 pH),115 ℃灭菌 15 min。 酵母粉和蛋白胨:购自OXIOD 公司;氨基酸标准品:购自Sigma 公司;其他化学试剂:均购自国药集团化学试剂有限公司;HPLC 级甲醇和乙腈: 购自苏州科盛公司;2×Taq PCR MasterMix:购自南京博尔迪生物科技有限公司;QuickCut DpnI 酶、PrimerStar DNA 聚合酶:购自 TaKaRa 公司;T4 DNA 连接酶、 反转录 cDNA 试剂盒: 购自 Fermentas 公司;T4 多聚核苷酸激酶:购自纽英伦生物技术 (北京) 有限公司;ChamQTMUniversal SYBR qPCR Master Mix: 购自诺唯赞生物;Simply P 总 RNA 提取试剂盒: 购自 Bioflux 公司;质粒提取试剂盒、细菌基因组提取试剂盒:购自天根生物科技有限公司;SanPrep 柱式PCR 产物纯化试剂盒、SanPrep 柱式DNA 胶回收试剂盒: 均购自生工生物工程股份有限公司。
1.2 实验方法
本研究所有用来构建质粒和突变菌的引物信息见表 1, 敲除突变菌株所用引物设计参照MG1655 基因组。 基因组和质粒的提取根据基因组和质粒提取试剂盒的说明书进行; 基因组片段用PrimerStar DNA 聚合酶 PCR 获得, 片段回收用SanPrep 柱式DNA 胶回收试剂盒,操作见说明书;菌落验证用 2×Taq PCR MasterMix 进行 PCR 验证。
以 MG1655 基因组为模板, 以引物 ptsHf1/ptsHr1 扩增出ptsH 基因上游片段ptsH-U, 以引物ptsHf2/ ptsHr2 扩增出ptsH 基因上游片段ptsH-D,再通过重叠PCR 把上下游片段重叠到一起得到ptsH 的敲除片段 ptsH-UD。 以 pTargetF 为模板,以引物 sgRNA-ptsH-F/T-sgRNA-R 扩增出构建pTargetF-ptsH 的片段, 片段经过 DpnI 酶消化,T4 Pnk 激酶磷酸化,T4 DNA 连接酶连接后化转到JM109 感受态中,构建和扩增质粒,在含50 mg/L 壮观霉素的LB 平板上生长10 h 后, 用引物Y-ptsHF/T-sgRNA-R 筛选正确的转化子, 得到敲除质粒pTargetF-ptsH。 100 ng 敲除质粒 pTargetF-ptsH 和500 ng 敲除片段 ptsH-UD 同时电转入 80 uL 的TWF001/pCas 电转感受态中,在 30 ℃、100 r/min 慢摇复苏2 h,涂在含50 mg/L 壮观霉素和卡那霉素的LB 平板,30 ℃培养 48 h 后用引物 ptsHf1/ ptsHr2 验证,得到正确敲除的转化子,正确的转化子转接含50 mg/L 卡那霉素和0.5 mmol/L 异丙基硫代半乳糖苷(IPTG)的 LB 试管诱导培养去除pTargetF-ptsH质 粒 ,42 ℃过 夜 培 养 去 除 pCas 质 粒 , 去 除pTargetF-ptsH 和pCas 质粒后分别在含壮观霉素和卡那霉素的LB 平板上进行反筛, 确定是否去除质粒成功,最后得到不含质粒的正确敲除ptsH 基因的突变菌TWF001ΔptsH。

表1 本研究中所用到菌株和质粒Table 1 Strains and plasmids used in the study
TWF001ΔptsG 的 构 建 过 程 同 上 述TWF001ΔptsH 的构建, 所用引物参照表 2 中的ptsGf1/ptsGr1、ptsGf1/ptsGr1、sgRNA -ptsG -F/T -sgRNA-R、Y-ptsG-F/T-sgRNA-R。
TWF001ΔptsH,Ptrc::PgalP 的 构 建 是 以MG1655 基因组为模板, 以引物galPf1/galPr1 扩增出galP 基因上游片段galP-U,以引物galPf2/galPr2扩增出galP 基因上游片段galP-D, 引物Trc-F/Trc-R 95 ℃变性、4 ℃退火形成trc 启动子片段,再通过重叠PCR 把上游、trc 启动子和下游片段重叠到一起得到galP 的敲入片段trc-galP。以pTargetF 为模板,以引物sgRNA-galP-F/T-sgRNA-R 扩增出构建pTargetF-galP 的片段, 片段经过 DpnI 酶消化,T4 Pnk 激酶磷酸化,T4 DNA 连接酶连接后化转到JM109 感受态中,构建和扩增质粒,在含50 mg/L 壮观霉素的LB 平板上生长10 h 后,用引物Y-galP -F/T-sgRNA-R 筛选正确的转化子, 得到敲除质粒pTargetF-galP。 100 ng 敲除质粒 pTargetF-galP 和500 ng 敲除片段 trc-galP 同时电转入 80 uL 的TWF001ΔptsH/pCas 电转感受态中,在 30 ℃、100 r/min慢摇复苏2 h,涂在含50 mg/L 壮观霉素和卡那霉素的 LB 平板,30 ℃培养 48 h 后用引物 Trc-F/galPr2验证,得到正确敲入的转化子,正确的转化子转接含50 mg/L 卡那霉素和0.5 mmol/L 异丙基硫代半乳糖苷(IPTG)的LB 试管诱导培养去除pTargetF-galP质 粒 ,42 ℃ 过 夜 培 养 去 除 pCas 质 粒 , 去 除pTargetF-galP 和pCas 质粒后分别在含壮观霉素和卡那霉素的LB 平板上进行反筛, 确定是否去除质粒成功, 最后得到不含质粒的正确敲入trc-galP 基因的突变菌 TWF001ΔptsH,Ptrc::PgalP。

表2 本研究构建质粒和菌株所用引物Table 2 Primers usedused to construct plasmids andstrains in this study

表3 本研究RT-PCR 所用引物Table 3 Primers used for RT-PCR in this study
1.3 发酵过程参数测定
1.3.1 生物量的测定 生长在LB 培养基中的种子液的OD600的测定:取1 mL 菌液,再用去离子水稀释相应的倍数充分混匀, 用紫外分光光度计测定600 nm 下的吸光度,再乘以相应倍数得到最后值。
生长在发酵培养基中的菌液的OD600的测定:取1 mL 菌液充分混匀,再用去1 mol/L 的稀盐酸稀释相应的倍数, 用紫外分光光度计测定在波长为600 nm 下的吸光度,再乘以相应倍数得到最后值。

图2 菌株构建流程和质粒图Fig. 2 Construction process of mutant strains and the map of plasmids
1.3.2 葡萄糖质量浓度的测定 发酵液取样,发酵液样品 12 000 r/min 离心, 取上清液 10 μL 用去离子稀释100 倍,用SBA40 还原糖测定仪测定葡萄糖的质量浓度。
1.3.3 氨基酸含量的测定
1) 样品预处理 发酵液12 000 r/min 离心,取500 μL 上清液加入到 500 μL 的 10%的三氯乙酸溶液中,充分混匀,4 ℃静置 4 h 以上。 12 000 r/min 离心15 min,用0.22 μm 的水相针式滤膜过滤上清液,上清液稀释合适的倍数后用HPLC 测定氨基酸含量。
2) 高效液相色谱 采用邻苯二甲醛柱前衍生法[18]。 仪器采用 Agilent 1200 超高效液相色谱仪,色谱柱为 Thermo ODS -2HYPERSIL C18 column(250 mm×4.0 mm,USA)。 流动相(水相):3.01 g 无水乙酸钠溶解于超纯水中,200 μL 三乙胺,5 mL 四氢呋喃,超纯水定容到1 L,乙酸调至pH 7.2。 洗脱相(有机相):3.01 g 无水乙酸钠溶解于200 mL 超纯水中,用5%醋酸调至pH 7.2,再加400 mL 的HPLC级甲醇和400 mL 的HPLC 级乙腈。
1.4 实时荧光定量PCR 分析
相关基因转录水平通过StepOnePlus 实时荧光定量仪(美国Applied Biosystems 公司)检测,所用引物见表2。发酵对数中期的发酵液,低速短暂离心取上清液去除发酵液中的CaCO3,12 000 r/min 离心收集菌体,RNA 的提取步骤、cDNA 的反转录合成以及RT-qPCR 的详细步骤及参数分别按照Bioflux RNA 提取试剂盒说明书、Fermentas 公司反转录cDNA 试剂盒和诺唯赞生物ChamQTM Universal SYBR qPCR Master Mix 说明书,数据的分析参照文献[19]。
2 结果与分析
2.1 突 变 菌 TWF001ΔptsH、TWF001ΔptsG 和TWF001ΔptsH,Ptrc::PgalP 的构建验证
3 株突变菌构建的电泳验证结果见图3。 对照菌 TWF001 用引物 ptsHf1/ptsHr2 进行 PCR 验证,扩增出1 500 bp 左右大小的条带, 如1 泳道所示,大小正确;突变菌TWF001ΔptsH 用引物ptsHf1/ptsHr2扩增出了1 200 bp 左右大小的条带, 如2 泳道所示,大小正确,电泳结果显示敲除了正确大小的条带,ptsH 基因敲除成功。

图 3 TWF001ΔptsH、TWF001ΔptsG 和 TWF001ΔptsH,Ptrc::PgalP 构建验证Fig. 3 Verification of TWF001ΔptsH,TWF001ΔptsG and TWF001ΔptsH,Ptrc::PgalP by PCR
突变菌 TWF001ΔptsG 用引物 ptsGf1/ptsGr2 PCR 验证,扩增出1 500 bp 左右大小的条带,如3泳道所示, 大小正确; 而对照菌TWF001 用引物ptsGf1/ptsGr22 扩增出的片段大小为 3 000 bp 左右,如4 泳道所示,大小正确。 电泳结果显示敲除了正确大小的条带,ptsG 基因敲除成功。
对照菌TWF001 经过基因组测序显示没有完整的galP 基因, 只存在galP 起始十几个碱基的片段,且没有其它同源性较高的序列。 要插入的片段包括一段galP 位置上的上游同源片段、trc 启动子和galP 基因,大小为2 048 bp,突变菌TWF001ΔptsH,Ptrc::PgalP 用引物 galPf1/galPr2 PCR 验证,扩增出2 000 bp 左右大小的条带, 如 5 泳道所示, 大小正确; 而 TWF001ΔptsH 基因组上没有引物 galPr2 结合位点, 所以扩增不出条带, 如6 泳道所示。TWF001 基因结构特殊,galP 基因本身有1 395 bp,在基因组上插入trc 启动子过表达galP 基因片段的难度较大。 PCR 验证正确的转化子,提取基因组,高保真酶扩增插入片段,对片段进行测序检验,完全正确插入片段并且没有突变的就是最终正确突变株。
2.2 TWF001ΔptsH 在不同葡萄糖质量浓度培养基中的摇瓶发酵结果分析
为了进一步提高TWF001 的L-苏氨酸含量,在TWF001 中 敲 除 ptsH 基 因 , 得 到 突 变 株TWF001ΔptsH。突变株在葡萄糖质量浓度为30 g/L[6]的培养基中进行摇瓶发酵,测定生长OD600、葡萄糖质量浓度和L-苏氨酸的产量。 为了探究L-苏氨酸产量和转化率能否随着葡萄糖质量浓度的升高进一步提升,又分别在含40、50、60 g/L 葡萄糖质量浓度的培养基中进行摇瓶发酵。
出发对照菌TWF001 和突变菌 TWF001ΔptsH在含 30、40、50、60 g/L 葡萄糖的培养基中的摇瓶发酵结果见图4。
由图 4(a)可知,TWF001ΔptsH 在各个葡萄糖质量浓度中的生长都要比TWF001 慢,生长受到明显的抑制。 当葡萄糖质量浓度为30 g/L 时,TWF001的最大OD600为15.59,当葡萄质量浓度继续升高到40、50 g/L 时,随着葡萄糖质量浓度的升高,TWF001的生长越好,60 g/L 的葡萄糖就开始抑制菌体的生长。 TWF001ΔptsH 在 30 g/L 葡萄糖中的最大 OD600为12.03,随着葡萄糖质量浓度升高到40、50、60 g/L时,虽然OD600变大了,但是发酵前期18 h 内,菌体生长非常缓慢,不利于发酵合成L-苏氨酸。
如图 4(b)所示,TWF001 在 30 g/L 葡萄糖质量浓度发酵时,发酵24 h 耗尽葡萄糖;随着葡萄糖质量浓度的升高, 发酵36 h 最多能耗掉50 g/L 的葡萄糖。TWF001ΔptsH 在30 g/L 葡萄糖质量浓度发酵时,发酵30 h 耗尽葡萄糖,随着葡萄糖质量浓度的升高,耗糖明显被抑制,发酵36 h 最多能耗掉40 g/L的糖, 并且当葡萄糖质量浓度等于或者高于40 g/L时,发酵前18 小时几乎不消耗葡萄糖,葡萄糖摄取被严重抑制,各个质量浓度中的耗糖速度都比对照菌慢。

图4 TWF001 和TWF001ΔptsH 在葡萄糖质量浓度为 30、40、50、60 g/L 的培养基中摇瓶发酵Fig. 4 Flask fermentation of TWF001 and TWF001ΔptsH in medium with 30,40,50,60 g/L glucose
如图 4(c)所示,TWF001 在 30 g/L 葡萄糖质量浓度发酵时L-苏氨酸产量为12.97 g/L, 糖酸转化率为0.43 g/g; 随着葡萄糖质量浓度升高到40 g/L时,L-苏氨酸产量为18.41 g/L, 此时达到最大转化率0.46 g/g;随着葡萄糖质量浓度升高到50 g/L 时,L-苏氨酸产量达到最大的22.56 g/L,转化率下降到0.45 g/g;当葡萄糖质量浓度为60 g/L 时,苏氨酸产量反而下降。TWF001ΔptsH 在30 g/L 葡萄糖发酵时L-苏氨酸产量为17.90 g/L, 比对照 TWF001 提高38.01%,这时的转化率为0.60 g/g;葡萄糖质量浓度升高到 40 g/L 时,L-苏氨酸产量达到最大的22.16 g/L,但是转化率下降到0.58 g/g,随着葡萄糖质量浓度升高到50、60 g/L 时, 苏氨酸产量反而下降,并且比对照菌要低。
ptsH 基因的敲除在30、40 g/L 葡萄糖质量浓度时能明显地提高L-苏氨酸的产量以及糖酸转化率,但是生长和糖耗受到抑制,并且40 g/L 的葡萄糖就开始明显抑制生长和糖耗,降低转化率,只适合在30 g/L 的葡萄糖进行发酵。
2.3 TWF001ΔptsG 在不同葡萄糖质量浓度培养基中的摇瓶发酵结果分析
为了进一步提高TWF001 的L-苏氨酸含量,在TWF001 中 敲 除 ptsG 基 因 , 得 到 突 变 株TWF001ΔptsG。 突变株在 30 g/L 葡萄糖的培养基中进行摇瓶发酵,测定生长OD600、葡萄糖质量浓度和L-苏氨酸的产量。 为了探究L-苏氨酸产量和转化率能否随着葡萄糖质量浓度的升高进一步提升,又分别在含40、50、60 g/L 葡萄糖的培养基中进行摇瓶发酵。
出发对照菌 TWF001 和突变菌TWF001ΔptsG在含 30、40、50、60 g/L 葡萄糖的培养基中的摇瓶发酵结果见图5。
如图 5(a)所示,TWF001ΔptsG 在各个葡萄糖质量浓度中的生长比TWF001 要慢一些,但生长没有受到太大的抑制。 当葡萄糖质量浓度为30 g/L 时,TWF001ΔptsG 的最大 OD600为 11.61,当葡萄糖质量浓度继续升高到40、50 g/L 时, 随着葡萄糖质量浓度的升高,菌体生长的越好,60 g/L 的葡萄糖在发酵前18 小时抑制菌体的生长。
如图 5(b)所示,TWF001ΔptsG 在 30 g/L 葡萄糖发酵时,发酵24 h 耗尽葡萄糖,与对照TWF001一致; 并且在40、50 g/L 的葡萄糖中的耗糖速度与对照菌十分接近,没有明显被抑制,当葡萄糖质量浓度升高到60 g/L 时,突变菌的葡萄糖摄取才表现出明显被抑制。
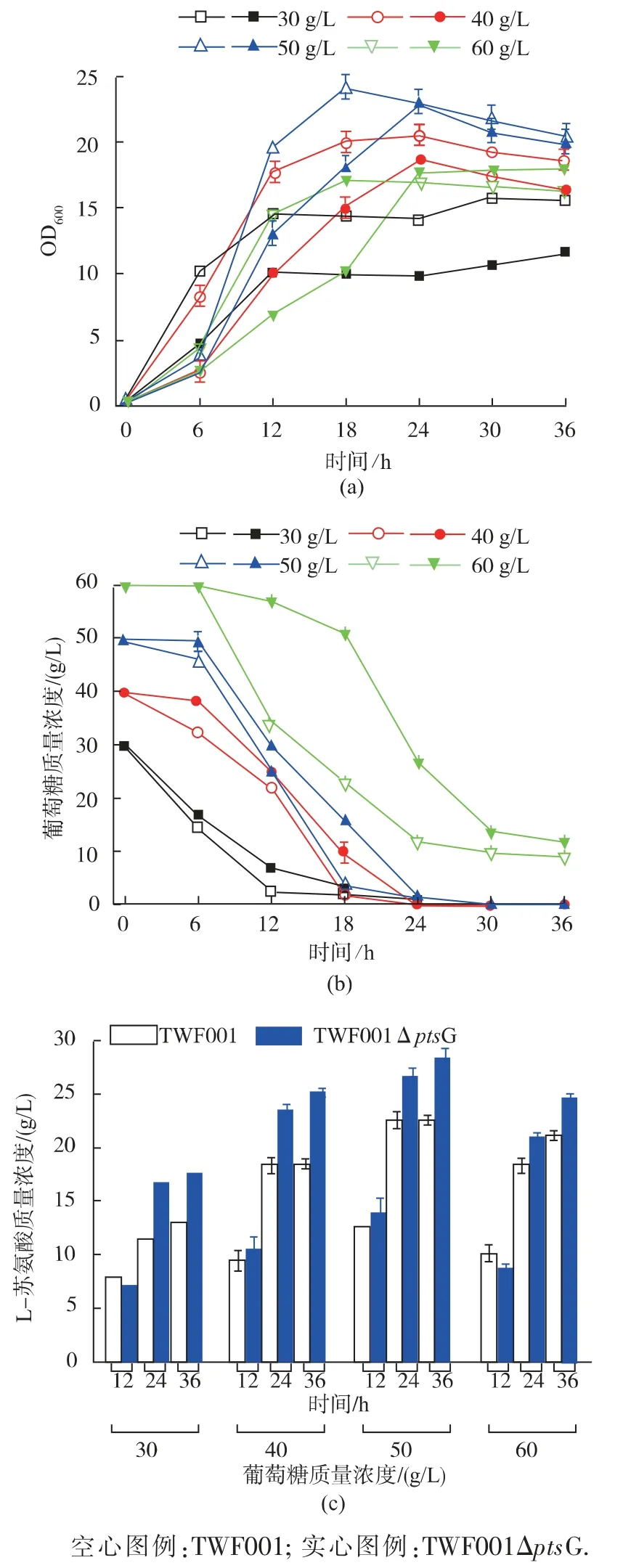
图5 TWF001 和TWF001ΔptsG 在葡萄糖质量浓度为30、40、50、60 g/L 的培养基中摇瓶发酵Fig. 5 Flask fermentation of TWF001 and TWF001ΔptsG in medium with 30,40,50,60 g/L glucose
如图 5(c)所示,TWF001ΔptsG 在 30 g/L 葡萄糖发酵时 L-苏氨酸产量为 17.59 g/L, 比对照TWF001 提高了35.62%,这时的转化率为0.59 g/g;当葡萄糖质量浓度升高到40 g/L 时,L-苏氨酸产量为25.23 g/L,比对照TWF001 提高37.05%,为这个菌的最高幅度,此时的转化率达到最大0.63 g/g;当葡萄糖质量浓度升高到50 g/L 时,L-苏氨酸产量达到最大的28.39 g/L,只比对照提高了25.84%,转化率也下降为0.57 g/g, 当葡萄糖质量浓度升高到60 g/L 时,苏氨酸产量反而下降。
ptsG 基因的敲除既能能明显提高L-苏氨酸的产量和糖酸转化率,40 g/L 葡萄糖时达到最大的转化率和L-苏氨酸产量提升幅度,是最适宜的葡萄糖质量浓度。
2.4 TWF001ΔptsH,Ptrc::PgalP 在不同葡萄糖质量浓度培养基中的摇瓶发酵结果分析
上述发酵结果显示,在30 g/L 葡萄糖培养基中进行发酵,TWF001ΔptsH 的L-苏氨酸产量为17.90 g/L,TWF001ΔptsG 的 L-苏氨酸产量为 17.59 g/L。为了进一步提高L-苏氨酸产量, 在TWF001ΔptsH的基因组上用trc 启动子过表达galP 基因, 得到突变株 TWF001ΔptsH,Ptrc::PgalP。 对突变株在 30 g/L葡萄糖的培养基中进行摇瓶发酵,为了探究L-苏氨酸产量和转化率能否随着葡萄糖质量浓度的升高进一步提升,又分别在含40、50、60 g/L 葡萄糖的培养基中进行摇瓶发酵。
出发对照菌TWF001 和突变菌TWF001ΔptsH,Ptrc::PgalP 在含 30、40、50、60 g/L 葡萄糖的培养基中的摇瓶发酵结果见图6。
《太原县志》中记载,明清时期风峪沟内水患严重,明洪武、嘉靖年间、清乾隆元年、十七年、三十三年、四十年均爆发过大规模的洪灾,对人畜、建筑、交通要道有较大的损害。在清乾隆四十一年完成对沙堰的修建之后,“工竣,而城北等村可永免山水冲突之患矣”[11]。从现存周家庄古村建筑建造手法及建筑材料可以看出,村中建筑呈现由高向低,由东向西发展的趋势,就是村民为防止水患危害,而采用的布局形式(图2)。
如图 6(a)所示,TWF001ΔptsH,Ptrc::PgalP 在30、50、60 g/L 葡萄糖中的生长比 TWF001 要慢一些,但生长没有受到太大的抑制。 当葡萄糖质量浓度为 30 g/L 时,TWF001ΔptsH,Ptrc::PgalP 的最大OD600为12.32;当葡萄糖质量浓度继续升高到40 g/L时,生长达到最好,并且比对照TWF001 略好。 升高到 50 g/L 时, 最大 OD600开始下降, 当继续升高到60 g/L 时,生长开始受到较明显的抑制。
如图 6(b)所示,TWF001ΔptsH,Ptrc::PgalP 在30 g/L 的葡萄糖中的耗糖速度与对照菌几乎一致,发酵18 h 耗尽30 g/L 的葡萄糖, 当葡萄糖质量浓度高于40 g/L 时,耗糖受到一定抑制,且质量浓度越高抑制越明显,发酵36 h,最多能耗尽40 g/L 的葡萄糖。

图 6 TWF001 和 TWF001ΔptsH,Ptrc::PgalP 在葡萄糖质量浓度为30、40、50、60 g/L 的培养基中摇瓶发酵情况Fig. 6 Flask fermentation of TWF001 and TWF001ΔptsH,Ptrc::PgalP in medium with 30,40,50,60 g/L glucose
如图 6(c)所示,TWF001ΔptsH,Ptrc::PgalP 在30 g/L 葡萄糖发酵时L-苏氨酸产量为17.66 g/L,比对照TWF001 提高了36.16%, 这时的转化率为0.60 g/g;当葡萄糖质量浓度升高到40 g/L 时,L-苏氨酸产量为 26.16 g/L, 比对照 TWF001 提高了42.10%,为3 株突变菌的最高幅度,此时的转化率达到最大的0.65 g/g,也是所有菌中最高的;当葡萄糖质量浓度升高到50、60 g/L 时,L-苏氨酸产量分别下降到到21.43、20.16 g/L。
ptsH 基因的敲除能提高L-苏氨酸的产量和糖酸转化率, 但是ptsH 的生长和糖耗受到明显的抑制,在敲除ptsH 突变菌的基础上用trc 启动子过表达galP 基因,不仅能改善生长和葡萄糖摄取[20],还能进一步提高L-苏氨酸的产量和糖酸转化率。
2.5 基因ptsH 和ptsG 的敲除以及galP 的过表达对L-苏氨酸合成相关基因的转录水平的影响
为了探究造成这3 株突变菌L-苏氨酸产量差异的原因,对大肠杆菌中影响L-苏氨酸合成的相关基因 thrA、thrB、thrC、pfkA、eno、pykA、gltA、gdhA 和aspC 进行了实时荧光定量PCR 分析。 如图7 所示,TWF001ΔptsH 中 thrA、thrB 和 thrC 的表达都有所提高,thrC 上升的最明显, 这可能是苏氨酸产量增加的主要原因之一;eno 基因明显提高有利于合成更多的PEP[21],pykA 基因的上调虽然不利于积累PEP,但有利于糖酵解的顺利进行合成丙酮酸,pfkA和gltA 基因是糖酵解和TCA 循环的关键限速步骤,它们的下调比较不利于生长[5],这可能是TWF001ΔptsH 生长和糖耗受到抑制的原因之一。TWF001ΔptsG 中 thrA 基因明显上调, 使得苏氨酸合成加强,pfkA 和eno 基因上调的比较明显, 这表示糖酵解的碳流和流向PEP 的碳流的提高,这有利于糖酵解和合成更多的PEP[11-13],更多的PEP 供应于苏氨酸的合成,pykA 基因的上调也使得生长更好,gdhA 基因的上调有利于合成更多谷氨酸, 而胞内氮源绝大部分来自谷氨酸[21],有利于L-苏氨酸的合成,apsC 基因的上调有利于合成天冬氨酸, 天冬氨酸是重要的苏氨酸前体物质。 在TWF001ΔptsH,Ptrc::PgalP 中,thrA、thrB、thrC、gdhA 和 aspC 这些基因都有明显的上调,这些基因的上调都直接有利于L-苏氨酸的合成, 这些可能是TWF001ΔptsH,Ptrc::PgalP 的L-苏氨酸产量的提升幅度和糖酸转化率最高的原因;eno 基因也明显上调有助于PEP的合成,但是pfkA 和gltA 基因上调较弱,不利于糖酵解和TCA 循环。
TWF001、TWF001ΔptsH 和 TWF001ΔptsG 中没有galP 基因,3 株菌中galP 的相对表达量相同,都为 1;TWF001ΔptsH,Ptrc::PgalP 中 galP 的相对表达量上升到16.2,提升幅度较大,表明插入基因组上的galP 基因得到表达。

图7 L-苏氨酸合成相关基因的实时荧光定量PCR 分析Fig. 7 RT -qPCR analysis of genes involved in L -threonien synthesis
3 结 语
在一株高产L-苏氨酸的大肠杆菌中分别单独敲除ptsH 和ptsG 基因,并在ptsH 敲除菌的基础上过表达galP 基因,进行摇瓶发酵,都能较大幅度提高L-苏氨酸产量和提高糖酸转化率,且菌株不携带任何表达质粒,有利于工业发酵。
ptsG 编码PTS 系统中特异识别结合葡萄糖的膜结合蛋白EIICBGlc,ptsH 编码PTS 中磷酸组氨酸搬运蛋白HPr,是转运磷酸基团过程的一种蛋白质,但是它为PTS 系统转运其他物质所共用。 敲除这两个基因都能使PTS 系统转运葡萄糖受阻,从而通过其他转运蛋白质如GalP 和MglABC 转运葡萄糖,能使用于PTS 系统转运葡萄糖消耗的PEP 积累而用于代谢合成如L-苏氨酸;RT-qPCR 也显示这两个基因的敲除能增加L-苏氨酸合成相关基因的表达,从而促进苏氨酸的合成。
对 TWF001、TWF001ΔptsH 和 TWF001ΔptsG进行摇瓶发酵, 此时葡萄糖质量浓度为30 g/L,TWF001ΔptsH 的生长和糖耗没有受到非常明显抑制, 此时它的L-苏氨酸产量和糖酸转化率是最高的, 为了进一步提高TWF001ΔptsH 的生长糖耗和L-苏氨酸产量,在基因组上用trc 过表达galP 基因。GalP 蛋白转运的葡萄糖只需要消耗一分子ATP,积累的PEP 可大部分用于合成代谢,而且能改善葡萄糖的摄取速度。 继续提升培养基中葡萄糖质量浓度,并进行摇瓶发酵,研究结果显示TWF001ΔptsH,Ptrc::PgalP 在40 g/L 的葡萄糖质量浓度时达到最大的L-苏氨酸产量26.16 g/L,提高幅度为42.11%,转化率能达到0.65 g/g。 先敲除PTS 系统中与转运葡萄糖相关的编码基因,使细菌不能以PTS 系统为主要的转运葡萄糖的途径,从而减少用于转运葡萄糖所需要的PEP,这样来自糖酵解的PEP 可大部分用于合成代谢, 再过表达GalP 这样不需要消耗其他重要中间代谢物的葡萄糖转运蛋白质,这样在不严重影响生长和糖耗的同时能大幅度提高L-苏氨酸产量和转化率。
之前有文献报道敲除ptsG 或者ptsH 基因可以用来生产其他物质[22-24],本试验在一株高产L-苏氨酸中只敲除ptsH、ptsG 以及过表达galP 就能较大幅度的提升产量和转化率,效果是十分明显的,这在工业应用上是比较有利的,但还可以进一步继续改造,使得生长和糖耗进一步提升。
因为本出发菌基因组结构较MG1655 复杂,生长缓慢、单菌落非常小,构建较困难,可以继续在本研究的基础上,在突变菌TWF001ΔptsG 中用trc 过表达 galP 基因, 以及在 TWF001ΔptsH,Ptrc::PgalP中过表达glk 基因,探究生长和L-苏氨酸产量情况并进行其他能提高苏氨酸产量的代谢改造。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
化学工业与工程(2022年1期)2022-03-29
昆明医科大学学报(2021年8期)2021-08-13
江西农业学报(2021年4期)2021-04-20
家庭百事通·健康一点通(2020年11期)2020-11-30
三农资讯半月报(2020年11期)2020-06-21
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
理科考试研究·高中(2014年8期)2014-10-17
