
微波辅助合成N-(2-嘧啶基)吲哚化合物
2020-11-09 13:42:18张首国林1
合成化学 2020年10期
朱 琳, 张首国, 彭 涛*, 王 林1,*
(1. 广东药科大学 药学院,广东 广州 510006; 2. 军事医学研究院 辐射医学研究所,北京 100850)
吲哚类化合物是分布最广泛的天然杂环化合物之一,具有多种生物活性,在医药、农业、化工等领域具有重要的应用价值[1-2]。由于吲哚电子分布的差别,酰化、炔化、烯基化等官能团化优先发生在吲哚的更活泼的C3位置,直接对吲哚的C—H键进行官能团化反应是一种获得吲哚衍生物的便捷途径[3-4]。近年来,通过过渡金属活化C—H键实现对吲哚C2位置选择性官能团化的反应越来越受到重视[2]。该方法首先在吲哚的氮原子上引入嘧啶导向基,嘧啶基团引导过渡金属催化剂活化C2位置的C—H键,进而实现C2位置的官能团化。目前,吲哚选择性C2官能团化反应中常用的过渡金属包括Rh、 Pd、 Cu、 Mn等,可方便地在C2位置引入醛基[5]、氯原子[6]、甲硫基[7]、苯甲酰基[8]、烷氧羰基[9]、氰基[10-11]、苯并噁唑[12]等基团。
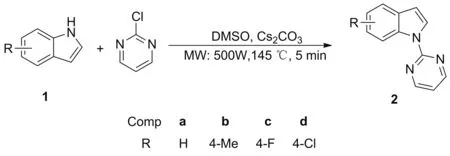
Scheme 1
N-(2-嘧啶基)吲哚类化合物是吲哚C2官能团化反应的重要原料,主要合成方法包括:2-氯嘧啶与吲哚为原料,氢化钠为碱,在DMF中于150 ℃反应12 h[12];在纳米氧化铜催化下,2-氯嘧啶与吲哚在碳酸钾-DMF中反应[13];在四(三苯基磷)钯催化下,2-溴嘧啶与吲哚硼酸酯在甲苯-水-乙醇混合液中发生C—N偶联反应[14]。上述方法需使用危险化学品、昂贵的纳米催化剂,且反应时间较长,不适用于N-(2-嘧啶基)吲哚化合物的快速合成。
微波辅助合成与传统合成方法相比,具有反应时间短、收率高、副反应少、合成效率高等优点[15]。鉴于此,本文开发了微波辅助合成N-(2-嘧啶基)吲哚化合物的方法,以取代吲哚和2-氯嘧啶为原料,在DMSO中以碳酸铯为碱,通过微波辅助合成在5 min内高收率地合成了4个N-(2-嘧啶基)吲哚化合物(2a~2d, Scheme 1),其结构经1H NMR和13C NMR 确证。
1 实验部分
1.1 仪器与试剂
XH100B型微波催化合成仪;YRT-3型熔点仪;Bruker 400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS 为内标)。
所用试剂均为化学纯或分析纯。
1.2 2a~2d的合成通法
将1a~1d5.0 mmol, 2-氯嘧啶7.5 mmol, DMSO 12 mL 和碳酸铯7.5 mmol加入反应瓶中,开启微波,搅拌下于500 W、 145 ℃反应5 min。冷却至室温,倾入100 mL水中,用乙酸乙酯(3×40 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸除溶剂,残余物经硅胶柱层析纯化得2a~2d。
N-(2-嘧啶基)吲哚(2a): 白色固体,收率95.4%, m.p.84~85 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 8.88(d,J=4.8 Hz, 2H), 8.75(dd,J=8.4 Hz, 0.8 Hz, 1H), 8.29(d,J=3.6 Hz, 1H), 7.69~7.63(m, 1H), 7.38~7.30(m, 2H), 7.23(td,J=7.5 Hz, 1.1 Hz, 1H), 6.80(dd,J=3.6 Hz, 0.8 Hz, 1H);13C NMR(DMSO, 101 MHz)δ: 159.40, 157.37, 135.13, 131.27, 126.16, 123.99, 122.51, 121.37, 117.68, 116.38, 107.26。
N-(2-嘧啶基)-4-甲基吲哚(2b): 白色固体,收率80.4%, m.p.114~116 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 8.87(d,J=4.8 Hz, 2H), 8.57(dd,J=8.4 Hz, 0.6 Hz, 1H), 8.28(d,J=3.7 Hz, 1H), 7.35(t,J=4.8 Hz, 1H), 7.22(dd,J=8.3 Hz, 7.3 Hz, 1H), 7.06~7.02(m, 1H), 6.84(dd,J=3.7 Hz, 0.8 Hz, 1H), 2.52(s, 3H);13C NMR(DMSO, 101 MHz)δ: 159.37, 157.39, 134.91, 130.87, 130.15, 125.55, 124.02, 122.82, 117.65, 114.00, 105.72, 18.75。
N-(2-嘧啶基)-4-氟吲哚(2c): 白色固体,收率85.2%, m.p.101~103 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 8.91(d,J=4.8 Hz, 2H), 8.59(d,J=8.4 Hz, 1H), 8.32(d,J=3.7 Hz, 1H), 7.42(t,J=4.8 Hz, 1H), 7.34(td,J=8.2 Hz, 5.7 Hz, 1H), 7.10~7.03(m, 1H), 6.88(dd,J=3.7 Hz, 0.6 Hz, 1H);13C NMR(DMSO, 101 MHz)δ: 159.52, 156.96(d,J=39.7 Hz), 154.33, 137.30(d,J=10.2 Hz), 126.70, 124.89(d,J=7.6 Hz), 119.63(d,J=22.3 Hz), 118.27, 112.93(d,J=3.5 Hz), 107.59(d,J=18.2 Hz), 102.26。
N-(2-嘧啶基)-4-氯吲哚(2d): 白色固体,收率81.5%, m.p.114~116 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 8.91(d,J=4.8 Hz, 2H), 8.72(ddd,J=7.3 Hz, 1.9 Hz, 0.5 Hz, 1H), 8.39(d,J=3.7 Hz, 1H), 7.42(t,J=4.8 Hz, 1H), 7.37~7.30(m, 2H), 6.84(dd,J=3.7 Hz, 0.5 Hz, 1H);13C NMR(DMSO, 101 MHz)δ: 159.53, 157.05, 135.86, 129.48, 127.38, 125.16, 125.02, 122.15, 118.34, 115.41, 104.71。

表1 反应条件对化合物2a收率的影响
2 结果与讨论
2.1 反应条件优化
以1a与2-氯嘧啶合成2a的反应为模板反应,对反应条件进行了优化。分别考察了碱、溶剂、反应时间、反应温度和微波功率对2a收率的影响,结果如表1。由表1可知,碱对反应具有重要影响,考虑到实验室使用的安全性和经济性,本实验将碱限定于氢氧化物和碳酸盐。对于多模常压微波合成仪而言,能达到的最高反应温度为溶剂的沸点,因此反应的溶剂本实验限定于沸点高、对微波吸收好的DMF和DMSO。
初始反应以氢氧化钾为碱、DMF为溶剂,于500 W、 145 ℃微波反应20 min,经分离纯化,2a收率为42.5%(Entry 1)。同样条件下,使用氢氧化钠为碱收率明显提高,为61.8%(Entry 2);使用碳酸钾收率降为29.6%(Entry 3);收率最高的为碳酸铯,可达72.7%(Entry 4)。固定碳酸铯为碱,为了增加碳酸铯的溶解性,使用DMF溶液为溶剂,收率降为25.8%(Entry 5)。使用DMSO为溶剂后,收率提高到96.0%(Entry 6)。将反应时间减少为10 min、 5 min,收率为95.7%、 95.4%(Entry 7, Entry 8),变化不明显;将反应温度升高至160℃,收率无明显变化,为95.2%(Entry 9),而降为130 ℃、120 ℃后,收率降为90.3%, 60.5%(Entry 10, Entry 11)。将反应温度固定为145 ℃,微波催化合成仪功率设定为600 W时,相应收率无显著差别,为95.8%(Entry 13),而设定为400 W时,发现反应液在5 min内,最高温度只能升至129.7 ℃,无法达到设定温度(Entry 12)。多模微波合成仪反应功率的设定与溶剂种类和体积等因素相关,在溶剂种类和体积确定的情况下,有一个最佳匹配功率,功率过低会导致升温速度较慢,功率过高会导致常压反应出现溶剂过热爆沸的现象。考虑收率和能源利用率等方面,确定最优的微波反应条件为:500 W、 145 ℃反应5 min。作为对比,采用传统油浴加热方式进行反应,反应60 min,收率87.4%(Entry 14)。多模微波合成仪的优点是反应腔体较大,适于放大合成,将反应投料量放大10倍(50 mmol),收率依然可以达到90.0%(Entry 15)。
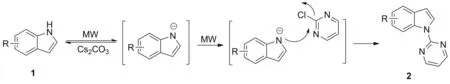
Scheme 2
2.2 底物拓展
在最佳微波反应条件下进行了底物拓展,结果表明,4-位取代的吲哚化合物均可与2-氯嘧啶顺利反应,收率大于80%。与无取代基的吲哚相比,4-位取代吲哚化合物的反应收率有所降低,其中4-位为吸电子取代基(氟)的底物,收率高于4-位为供电子取代基(甲基)的底物。
2.3 反应机理
根据实验结果,提出可能的反应机理如Scheme 2所示:吲哚化合物在碳酸铯的作用下脱去氮上的氢原子生成氮负离子,这是一个酸碱平衡反应,由于微波的电磁场效应使得氮负离子的稳定性增强,平衡更多的向右移动。氮负离子再与2-氯嘧啶发生亲核取代反应得到产物,在这一步反应中微波的热效应可以加速反应的进行。微波通过电磁场效应和热效应,使微波辅助的N-(2-嘧啶基)吲哚化合物的合成速率和收率均优于传统合成方法。
以吲哚、取代吲哚和2-氯嘧啶为原料,通过微波辅助合成了4个N-(2-嘧啶基)吲哚化合物。与已报道的方法相比,该方法具有安全性好、成本较低、反应时间短、收率较高等优点。
猜你喜欢
中学生数理化·自主招生(2024年6期)2024-06-24 11:15:29
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
昆明医科大学学报(2021年8期)2021-08-13 08:59:04
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21 07:51:12
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
武警医学(2018年10期)2018-11-06 07:04:34
当代化工研究(2016年6期)2016-03-20 16:21:42
材料科学与工程学报(2016年5期)2016-02-27 07:11:31
