
回溯搜索丁型肝炎病毒核酶的折叠路径
2020-11-06 03:30邹彦娟张小龙
萍乡学院学报 2020年3期
邹彦娟,张小龙,廖 昆
回溯搜索丁型肝炎病毒核酶的折叠路径
邹彦娟,张小龙,廖 昆
(萍乡学院 机械电子工程学院,江西 萍乡 337000)
RNA生物功能与其折叠的动力学过程密切相关。文章采用枚举法给出丁型肝炎病毒(Hepatitis Delta Virus, HDV)核酶序列的折叠空间,作为回溯搜索的解空间。结合自由能曲面计算了空间内构象之间的转换速率。采用回溯搜索算法,从天然态开始确定了HDV核酶序列的折叠路径,并利用主方程方法分析了路径上具有直接转换关系的结构之间占据几率随时间的变化。结果显示,HDV天然态的形成过程呈两相性,并且主要有3条折叠路径。另外在折叠过程中存在一个滞留时间较长的中间态假结结构。
折叠路径;回溯搜索;主方程方法;枚举法;丁型肝炎病毒核酶
RNA生物功能的实现与其折叠过程中形成的结构有关。RNA在折叠过程中可能会沿着不同的路径形成复杂的二级或三级结构。目前了解RNA结构信息实验的方法有X射线晶体衍射方法术[1~3]、核磁共振法[4]和冷冻电镜技术[5~6]等。这些实验方法测量结果准确,可以观察到RNA的晶体结构及其在折叠过程中所经历的中间态,但耗时长,成本高。随着计算机的发展和应用,通过理论计算模拟,如动态规划算法[7]、分子动力学模拟算法[8]、蒙特卡洛算法[9]等,预测RNA的动力学过程成为研究RNA的一个捷径。但RNA的空间结构数目会随着链长呈指数增长[10],利用计算机很难穷尽所有的构象取样。为此,笔者通过C语言编程,在个人计算机上运行回溯搜索RNA酶的折叠路径,从而可以分析折叠过程中的主要结构及其对折叠过程造成的影响。
回溯搜索算法是程序设计中一种重要的基础算法,是搜索算法中的一种控制策略。回溯搜索算法的基本原理是在搜索过程中从某一条搜索路径一直往前走,直到不能前进,然后退回来选择另一条路径再走。利用回溯搜索算法解决问题,首先要定义问题的解空间,然后利用便于搜索的方法组织解空间,并从问题的解空间中搜索得到可行解。基于RNA结构碱基配对关系,可以将回溯算法应用于寻找RNA折叠过程任意两态之间的折叠路径。
丁型肝炎病毒(Hepatitis Delta Virus, HDV)核酶是乙型肝炎病毒(Hepatitis B Virus, HBV)的单链RNA病毒,在细胞内通过双滚环(double rolling-circle)机制完成复制,新产生的RNA再由基因或反基因酶剪切成单体术[11~12]。Perrotta和Been在实验中采用化学电泳法对样品分析发现,正义和反义HDV序列在包含85个核苷酸时自剪切活性最佳术[13~14]。Bevilacqua实验室对HDV核酶的研究发现,HDV核酶的折叠路径受到假结(Pseudoknot)的影响,其折叠动力学过程呈两相性[15](折叠路径分成了两条)。理论研究方面,Chen等人发现,HDV核酶在折叠过程中由于假结的影响存在快慢两条路径[16]。
本文根据RNA碱基配对规则,采用枚举法定义HDV核酶的构象空间,作为回溯搜索的解空间;根据RNA折叠特点结合自由能曲面计算了构象之间的转换速率,作为回溯搜索条件;利用主方程方法术[17~18]计算各个构象的占据几率分布,分析HDV序列的折叠特点;最后回溯搜索计算了HDV序列由展开链到天然态结构的折叠路径,找出了折叠过程中出现的重要的中间态结构,并分析了其在折叠过程中发挥的作用。研究结果显示,HDV序列折叠过程存在3条主要的路径:一条快速路径,两条慢速路径。
1 研究方法
1.1 枚举生成构象回溯空间
RNA分子在细胞内通过形成连续的碱基配对可以折叠形成稳定的螺旋结构。笔者以螺旋为基本单元构建RNA结构,并限制每个螺旋至少由三个连续的碱基配对构成。在程序中,首先,对RNA序列进行顺序搜索,生成所有可能的螺旋组成螺旋数据库。然后,确定不同螺旋之间的相互关系(图1)。螺旋之间存在三种关系:(a)完全兼容:两个螺旋可以共存,无相互交叠的碱基;(b)完全不兼容:两个螺旋不能共存,碱基完全交叠;(c)部分兼容:两个螺旋可以部分共存,碱基部分交叠。最后,选取完全兼容或部分兼容的螺旋组装RNA结构,生成构象空间。

图1 螺旋之间的相互关系
1.2 计算构象之间的转换速率
在以螺旋为基本单元的模型中,RNA结构之间的跃迁,通过螺旋的形成、断开和交换来进行。
(1)形成螺旋的速率。RNA形成一个新的螺旋可从任意一个“碱基堆积”开始,不同的“碱基堆积”对应多条不同的生长路径,如图2(a)所示。对于任意一个“碱基堆积”其相邻的碱基形成速率要远远快于其他碱基的形成,且其自由能曲面呈逐渐下降的趋势,如图2(b)所示。对于含有三个连续碱基堆积的螺旋,其稳定性足以抵抗环境的热扰动,所以计算某条路径的折叠速率可用形成前三个碱基堆积的速率进行估算术[19~20],计算公式是:


(3)螺旋交换速率。如果A与B两个螺旋相互交叠,则通过螺旋A断开、螺旋B再形成的自由能势垒的高度,要高于螺旋A断开的同时螺旋B形成造成的自由能势垒的高度。可根据隧道路径理论计算两个螺旋之间的交换速率(如图2(c)所示):

式中,和分别表示碱基在螺旋A中形成(在螺旋B中打开)或碱基在螺旋B中形成(在螺旋A中打开)的速率,为玻尔兹曼常数,为螺旋A与螺旋B所在RNA结构的自由能差值。
1.3 计算构象的占据几率

1.4 回溯搜索折叠路径

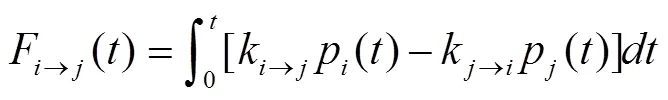
2 计算结果
2.1 构象占据几率的变化
本文用于计算的HDV序列包含95个核苷酸(CCUGAUGGCCGGCAUGGUCCCAGCCUCCUCGCUGGCGCCGGCUGGGCAACAUUCCGAGGGGACCGUCCCCUCGGUAAUGGCGAAUGGGACGCACA),生成的构象空间包含2971个RNA结构。通过计算构象空间中各个态占据几率随时间的变化,发现在折叠过程中起重要作用的中间态结构“1627”,在反应开始后的10-3.5s内,大约有50%的HDV序列快速的形成结构“1627”,在10-3.5s到10 s内HDV序列形成结构“1627”的速率变得缓慢,10 s到1000 s的时间内,结构“1627”缓慢地转换成其他结构,如图3(a)所示。我们同时发现天然态结构的形成明显的分为两个阶段,在折叠开始的18s内大约有40%的HDV序列迅速的形成天然态结构,剩余的大约60%的RNA序列在随后的35分钟内逐渐形成天然态结构,如图3(b)所示。造成天然态结构的形成分成两个阶段的原因可能是“1627”在10 s前快速形成,在10s后慢速形成,为了解释这一原因本文对HDV的折叠路径进行搜索。
(a)HDV序列折叠动力学过程,“1”是展开链,“1627”是一中间态,“Native”是天然态结构;(b)天然态结构占据几率随时间变化分成两个阶段
图3 HDV序列的中间态和天然态的占据几率随时间的变化
2.2 回溯搜索HDV序列的折叠路径
通过计算HDV序列构象空间内各RNA结构占据几率随时间的变化,可知HDV序列在折叠过程中存在滞留时间较长的中间态“1627”。为了解释“1627”在折叠过程中发挥的作用,本文采用回溯搜索算法,计算了HDV序列的折叠路径,同时计算了在折叠路径上有直接转换关系的主要结构之间的转换流量,如图4(a)、(b)、(c)所示。
HDV天然态结构由构象“1175”与构象“1636”转换形成,分别约占87%,5%,见图4(a)。构象“1175”与天然态结构之间的转换呈现出了二相性,见图4(a),约40%的HDV序列在折叠开始的18s内快速转换成天然态结构,而约50%的HDV序列在35 min内缓慢地转换成为天然态结构,这与天然态结构的形成分成两个阶段相一致。


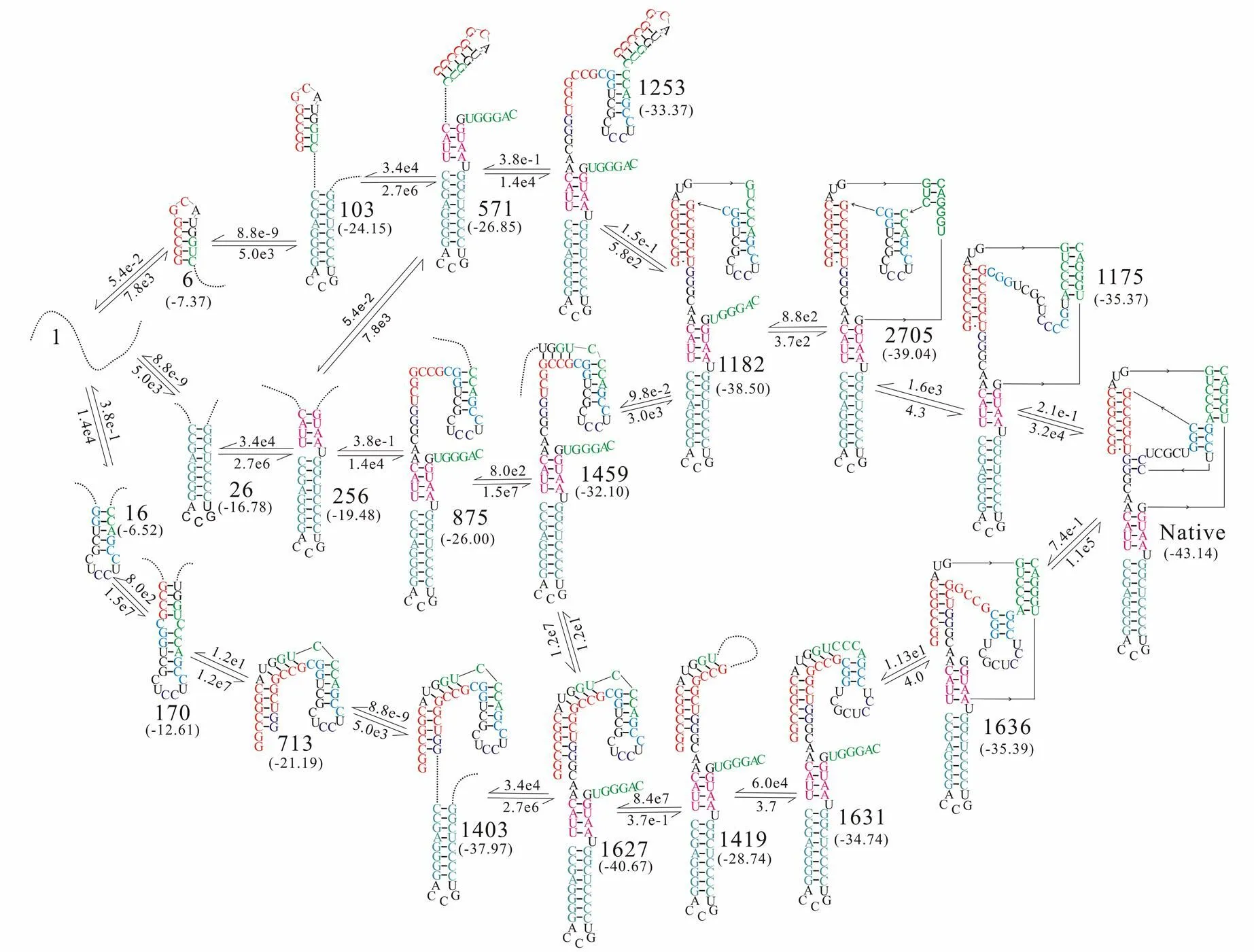
HDV序列从展开链到天然态的主要折叠路径,沿箭头方向的数字为转换速率(单位:s-1),括号中的数字为构象自由能(单位:kcal/mol)
结论
本文利用回溯搜索算法,通过搜索HDV核酶由展开链形成天然态结构的主要路径,很好地解释了HDV折叠过程中分成两个阶段的原因。同时计算结果显示,在HDV序列折叠过程存在三条主要的折叠路径。从中间态结构“1182”开始,转换过程分成了快慢两个阶段,对应构象“1253”的路径为快速路径,对应构象“1459”的路径为慢速路径。导致构象“1459”转换形成构象“1182”较慢的原因是,在折叠过程中大约有50%的HDV序列在构象“1627”滞留了约100s的时间,之后这些构象才在约35 min的时间内缓慢转换形成构象“1459”。本研究进一步的工作,可将回溯算法与其他算法结合,不断改进回溯算法,更好的预测RNA序列的折叠过程。
[1] TERESHKO V, WALLACE ST, USMAN N, et al. X-ray crystallographic observation of “ in-line ” and “ adjacent ” conformations in a bulged self-cleaving RNA / DNA hybrid[J]. RNA, 2001, 7(3): 405~420.
[2] ROBERT R. CRICHTON, RICARDO O. LOURO. Practical approaches to biological inorganic chemistry (Second Edition)[M]. Holland: Elsevier, 2020: 375~416.
[3] JUN S, HIRATA A, KANAI T, et al. The X-ray crystal structure of the euryarchaeal RNA polymerase in an open-clamp configuration[J]. Nat Commun, 2014,5(5132): 1~11.
[4] FÜRTIG B, RICHTER C, WÖHNERT J, et al. NMR spectroscopy of RNA[J]. ChemBioChem, 2003, 4(10): 936~962.
[5] BARRAUD P, GATO A, HEISS M, et al.Time-resolved NMR monitoring of tRNA maturation[J]. Nature Communications, 2019, 10(1): 3373.
[6] GARMANN RF, GOPAL A, ATHAVALE SS, et al. Visualizing the global secondary structure of a viral RNA genome with cryo-electron microscopy[J]. RNA, 2015, 21(5): 877~886.
[7] AKUTSU T. Dynamic programming algorithms for RNA secondary structure prediction with pseudoknots[J]. Discret Appl Math, 2000, 104(1~3): 45~62.
[8] HASHEM Y, AUFFINGER P. A short guide for molecular dynamics simulations of RNA systems[J]. Methods, 2009, 47(3): 187~197.
[9] CLOTE P, BAYEGAN AH. Mathematical Biology RNA folding kinetics using Monte Carlo and Gillespie algorithms[J]. J Math Biol, 2017, 76(5): 1195~1227.
[10] 邹彦娟, 石方. 在直角坐标系中回溯枚举RNA结构[J]. 萍乡学院学报, 2018, 35(6): 57~60.
[11] MACNAUGHTON TB, SHI ST, MODAHL LE, et al. Rolling circle replication of Hepatitis Delta Virus RNA is carried out by two different cellular RNA polymerases[J]. Virology, 2002, 76(8): 3920~3927.
[12] BROWN TS, CHADALAVADA DM, BEVILACQUA PC. Design of a highly reactive HDV ribozyme sequence uncovers facilitation of RNA folding by alternative pairings and physiological ionic strength[J]. J Mol Biol, 2004, 341(3): 695~712.
[13] PERROTTA AT, BEEN MD. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA[J]. Nature, 1991, 350(6317): 434~436.
[14] PERROTTA AT, MICHAEL DB. The self-cleaving domain from the genomic RNA of hepatitis delta virus: sequence requirements and the effects of denaturant[J]. Nucl Acids Res, 1990, 18(23): 6821~6827.
[15] CHADALAVADA DM, SENCHAK SE, BEVILACQUA PC. The folding pathway of the genomic hepatitis delta virus ribozyme is dominated by slow folding of the pseudoknots[J]. J Mol Biol, 2002, 317(4): 559-575.
[16] CHEN JW, GONG S, WANG YJ, et al. Kinetic partitioning mechanism of HDV ribozyme folding[J]. J Chem Phys, 2014, 140(2): 025102.
[17] ZHAO PN, ZHANG WB, CHEN SJ. Cotranscriptional folding kinetics of ribonucleic acid secondary structures[J]. J Chem Phys, 2011, 135(24): 245101.
[18] ZHANG WB, CHEN SJ. Master equation approach to finding the rate-limiting steps in biopolymer folding[J]. J Chem Phys, 2003, 118(7): 3413~3420.
[19] GONG S, WANG YJ, ZHANG WB. The regulation mechanism of yitJ and metF riboswitches[J]. J Chem Phys, 2015, 143(4): 045103.
[20] GONG S, WANG YJ, ZHANG WB. Kinetic regulation mechanism of pbuE riboswitch[J]. J Chem Phys, 2015, 142(1): 015103.
[21] ZHAO PN, ZHANG WB, CHEN SJ. Predicting secondary structural folding kinetics for nucleic acids[J]. Biophys J, 2010, 98(8): 1617~1625.
Backtracking Search for the Folding Pathway of the Hepatitis Delta Virus Ribozyme
ZOU Yan-juan, ZHANG Xiao-long, LIAO Kun
(School of Mechanical and Electronic Engineering, Pingxiang University, Pingxiang Jiangxi 337000, China)
RNA biological functions are closely related to its folding kinetics. In the paper, the folding space of the Hepatitis Delta Virus (HDV) ribozyme sequence is given by enumeration method as a solution space for backtracking search. The conversion rates between conformations within the space were calculated in combination with free energy surfaces. The backtracking search algorithm was used to determine the folding path of the HDV ribozyme sequence from the native state, and the change in the occupancy probability over time between structures with direct transition relationships in the path was analyzed by the master equation method. The results show that the natural state formation process of HDV is biphasic and that there are three main folding paths. In addition, one pseudoknot trap state is formed with a long residence time in the folding process.
folding path; backtracking search; master equation method; enumeration; HDV ribozyme
2020-03-07
萍乡学院青年科研基金(2018D0227、2018D0228)
邹彦娟(1988—),女,河南濮阳人,讲师,硕士,研究方向:凝聚态物理学的研究。
O469;Q61
A
2095-9249(2020)03-0079-06
〔责任编校:吴侃民〕
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11
舰船科学技术(2022年11期)2022-07-15
中国典型病例大全(2022年13期)2022-05-10
中国典型病例大全(2022年9期)2022-04-19
科学之谜(2021年2期)2021-04-25
军民两用技术与产品(2021年2期)2021-04-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
科学导报(2020年54期)2020-09-09
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
