
HPLC法测定盐酸莫西沙星片中光降解杂质的含量
2020-11-03 02:11李林梅宋太发索成林胡雄唐娟丽李帅帅
邵阳学院学报(自然科学版) 2020年5期
李林梅,宋太发,索成林,胡雄,唐娟丽,李帅帅
(湖南天济草堂制药股份有限公司,湖南 长沙,410205)
盐酸莫西沙星由德国拜耳公司于20世纪90年代末期开发上市的第4代广谱氟喹诺酮类抗菌药,自1999年以来已在多个国家上市,盐酸莫西沙星片于2002年在中国批准上市,上市剂型有片剂、小容量注射剂及大容量注射剂。
朱荣等[1-2]采用梯度洗脱的方法,对盐酸莫西沙星含量和有关物质的检测方法进行了研究,其中包括杂质A~E。苏敏等[3]采用等度洗脱,通过流动相比例优化,实现了盐酸莫西沙星葡萄糖溶液中杂质A~E的分离检测。高宏伟等[4-5]也采用高效液相色谱法,对盐酸莫西沙星杂质进行了分析。董雅等[6]对莫西沙星有关物质研究的相关文献进行了综述报道,文章指出目前针对莫西沙星有关物质的控制,大多仅研究了杂质A~E。郑家晴等[7]报道了A~F的有关物质分析方法。
2012年,URSZULA等[8]报道了莫西沙星在紫外照射条件下,以及有无Cu(Ⅱ),Zn(Ⅱ),Al(Ⅲ)和Fe(Ⅲ)等离子的溶液和固体中,莫西沙星会发生光降解,其中2’-氧代莫西沙星为光降解杂质之一。本文结合盐酸莫西沙星的生产工艺路线,针对6个杂质,分别为1-环丙基-6,7-二氟-1,4-二氢-8-羟基-4-氧代-3-喹啉羧酸(杂质K,中间体降解产物)、1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸乙酯(杂质L,工艺杂质)、6-羟基莫西沙星(杂质N,工艺杂质)、2’-氧代莫西沙星(杂质M,文献报道的光降解杂质)、加替沙星中间体(母核,合成莫西沙星的关键物料)、1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸-(O3,O4)-双-(乙酰氧基)-硼酸酯(中间体,工艺杂质),进行有关物质分析方法开发。同时杂质A~F在该方法下进行了专属性研究,排除杂质A~F不会干扰这6个杂质的检出。
对各国收录的盐酸莫西沙星片标准比较,发现其有关物质检测方法大多采用苯基柱。本文优化了有关物质检测方法,采用常用的C18柱和等度洗脱的方法,可有效分离上述6个杂质。同时对方法进行验证,结果显示:此方法专属性好,操作简便,对多个杂质均能有效分离,可作为有关物质检查方法。
1 材料与方法
1.1 仪器与试剂
Ultimate 3000高效液相色谱仪(Thermo Fisher Scientific公司),EX125DZH型电子天平(OHAUS公司,精密度1/100 000)。
盐酸莫西沙星片,批号190301,190302(湖南天济草堂制药股份有限公司);杂质A,批号151023(北京康派森医药科技有限公司,含量99.4%);杂质B,批号33266(北京康派森医药科技有限公司,含量99.2%);杂质C,批号34835(北京康派森医药科技有限公司,含量99.4%);杂质D,批号27546(北京康派森医药科技有限公司,含量99.6%);杂质E,批号39081(北京康派森医药科技有限公司,含量98.3%);杂质F,批号25520(北京康派森医药科技有限公司,含量99.9%);杂质N,批号MX-190422(深圳博泰尔生物技术有限公司,含量96.1%);中间体,批号39065(北京康派森医药科技有限公司,含量99.9%);杂质M,批号2798-072A3(TLC,含量99.4%);杂质L,批号19-MVI-71-2(TRC,含量98.0%);杂质K,批号MXJ20111201(北京康派森医药科技有限公司,含量99.3%);母核,批号MXG20111201(北京康派森医药科技有限公司,含量98.4%);盐酸莫西沙星对照品,批号3.0(EP,含量96.9%);甲醇、乙腈为色谱纯,水为超纯水(Millipore公司),其余试剂均为分析纯。
1.2 方法
1.2.1 色谱条件
采用高级液相色谱法(HPLC)进行检测,色谱条件为:色谱柱为Agilent poroshell EC-C18柱(5.0 μm,4.6 mm×250 mm,Agilent公司);四丁基硫酸氢铵缓冲液∶甲醇(90∶10)(V/V)为流动相A;甲醇∶四丁基硫酸氢铵缓冲液(90∶10)(V/V)为流动相B;流动相为A稀释溶剂;采用等度洗脱,检测波长为247 nm;柱温30 ℃;进样体积20 μL;流速1.3 mL/min。
1.2.2 杂质结构
盐酸莫西沙星涉及的6个杂质,结构见图1。
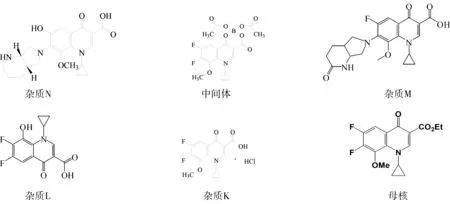
图1 盐酸莫西沙星6个杂质结构Fig.1 The six impurity Structrue of Moxifloxacin hydrochloride
1.2.3 专属性
杂质定位溶液制备:精密称取A~F等各杂质适量,制成100 μg/mL的各杂质贮备液;精密移取1 mL各杂质贮备液,分别置100 mL量瓶中,稀释定容,制成1.0 μg/mL的单个杂质定位溶液。
1.2.4 系统适用性溶液制备
精密称取杂质K,L,M,N,中间体和母核等各杂质适量,制成100 μg/mL的单个杂质贮备液。精密移取1 mL各杂质贮备液,置同一100 mL量瓶中,稀释定容,制成终质量浓度约为1.0 μg/mL的杂质混合溶液;另精密称取盐酸莫西沙星对照品适量,置10 mL量瓶中,加杂质混合溶液稀释并定容至刻度,制成各杂质质量浓度为1 μg/mL、莫西沙星约为1.0 mg/mL的系统适用性溶液。
1.2.5 系统适用性试验
将系统适用性溶液连续进样6次,进样体积20 μL,系统适用性要求:主峰莫西沙星与相邻杂质,以及各杂质之间的分离度均不得小于1.5,主峰理论板数不得小于20 000;杂质峰面积的相对标准偏差(relative standard deviation,RSD)均不得小于2.0%,保留时间的RSD均不得小于1.0%(n=6)。
1.2.6 供试品溶液的制备
精密称取盐酸莫西沙星片适量(相当于莫西沙星50 mg),置50 mL量瓶中,加稀释剂溶解并稀释至刻度,摇匀,得1.0 mg/mL的供试品溶液。
2 结果
2.1 方法专属性
空白溶剂、各杂质定位溶液、系统适用性溶液等样品溶液,进样体积20 μL,注入液相色谱仪,记录色谱图,各杂质不干扰2’-氧代莫西沙星的测定,试验结果见表1,相关图谱见图2和图3。

表1 方法专属性结果Table 1 Resuit of method specificity

图2 空白溶剂图谱Fig.2 Chromatogram of blank solution

图3 系统适用性溶液图谱Fig.3 Chromatogram of system suitability solution
2.2 进样精密度
按所述色谱条件试验,精密量取系统适用性溶液20 μL,注入色谱仪,记录色谱图,重复进样6次。2’-氧代莫西沙星峰保留时间RSD为0.10%;峰面积RSD为1.18%,说明仪器精密度良好。
2.3 定量限与检测限
同2.2项操作,精密量取2’-氧代莫西沙星(杂质M)对照贮备液适量,用稀释剂逐级稀释,分别注入色谱仪,当信噪比为10∶1时,得定量限(LOQ),连续进样6次,记录色谱图。当信噪比为3∶1时,得检测限(LOD),连续进样3次,记录色谱图,定量限为3.021 ng,RSD%(n=6)4.56%,检测限为1.510 ng。
2.4 线性与范围
精密量取2’-氧代莫西沙星(杂质M)对照贮备液适量,稀释成0.015 1,0.201 0,0.503 0,0.806 0,1.007 0和2.517 0 μg/mL的系列质量浓度线性溶液,各质量浓度进样20 μL,以2’-氧代莫西沙星的质量浓度与峰面积之比进行线性回归。得回归方法:y=0.297x-0.016(r=0.999 5),线性范围为0.151~2.517 μg/mL,定量下限为0.151 μg/mL。
2.5 溶液稳定性
取2.3项下2’-氧代莫西沙星(杂质M)对照贮备液适量,稀释制成终质量浓度为1.0 μg/mL的对照品溶液,室温放置,分别于0,4,12,14,16,18和20 h精密量取20 μL,测定峰面积,峰面积RSD(n=6)为1.17%,小于5%,说明对照品溶液室温0~20 h内稳定。
2.6 重复性
取盐酸莫西沙星适量,用稀释剂溶解并稀释制成每1 mL中约含1 mg的溶液,平行配制6份,按照2’-氧代莫西沙星检测方法测定,6份供试品中均未检出2’-氧代莫西沙星,结合回收率结果,该法重复性良好。
2.7 回收率
配制低、中、高3个质量浓度2’-氧代莫西沙星(5.04,10.12和15.03 μg/mL)标准系列样品各3份,进样20 μL,记录2’-氧代莫西沙星的峰面积。按照2.3项下新配制2’-氧代莫西沙星对照品溶液,终质量浓度为1.0 μg/mL,进样20 μL,记录对照峰面积。2’-氧代莫西沙星3个质量浓度水平的回收率为97.7%,98.5%和99.7%,平均回收率98.6%,RSD(n=9)为1.51%,结果见表2。

表2 2’-氧代莫西沙星回收率结果(n=9)Table 2 Recovery results of 2’-oxymoxifloxacin in moxifloxacin hydrochloride (n=9)
3 讨论
目前未见相关文献报道盐酸莫西沙星片中光降解杂质2’-氧代莫西沙星的测定方法,本文建立的HPLC方法,不仅能有效分离多个杂质,同时能有效检测2’-氧代莫西沙星,专属性良好。在建立本定量分析方法时,对流动相比例、检测波长、流速、柱温等分析方法进行了筛选优化,改善了各杂质的分离度,最终确定本文采用的色谱条件。
采用Agilent Poroshell EC-C18色谱柱,固定柱温35 ℃,流速1.3 mL/min,检测波长设为293 nm[1](293 nm为莫西沙星的最大吸收波长),通过调整流动相A相和B相的比例进行检测方法的优化。当流动相比例为60∶40(V/V)时,运行时间60 min内杂质K,L,M,中间体和母核5个杂质未完全分离,其中2个杂质相互重叠为一个峰;当调整流动相比例至(50∶50)(V/V)时,运行时间35 min内5个杂质均能有效分离,最小分离度为1.45,各峰峰行良好。但该检测条件,杂质N未出峰。
鉴于杂质N未出峰,通过对单个杂质溶液、混合杂质溶液的紫外吸收波长扫描分析,最终选用247 nm作为该方法的检测波长。固定流动相比例至50∶50(V/V),在247 nm进行HPLC分析,结果显示:杂质K,L,M,N,中间体和母核6个杂质与主峰全部实现有效分离,最小分离度为2.97,各峰峰行良好。
将流速设为1.0 mL/min,杂质K,L,M,N,中间体和母核6个杂质全部有效分离,最小分离度达1.81,各峰峰行良好,说明流速的调整,对分离效果影响不大。
将波长设为247 nm,柱温设为30 ℃,杂质K,L,M,N,中间体和母核6个杂质全部有效分离,最小分离度达3.21,各峰峰行良好,说明降低柱温,可提高分离效果。
综上,采用Agilent Poroshell EC-C18色谱柱,四丁基硫酸氢铵缓冲液∶甲醇(90∶10)(V/V)为流动相A;甲醇∶四丁基硫酸氢铵缓冲液(90∶10)(V/V)为流动相B;流动相A为稀释溶剂;采用等度洗脱,检测波长为247 nm;柱温30 ℃;进样体积20 μL;流速1.3 mL/min。参照相关文献[9-10]报道的验证思路,结果表明:本文研究方法专属性好,灵敏度满足检测要求,可作为新的分析方法,用于盐酸莫西沙星片多个杂质的研究。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
艺术品鉴(2020年6期)2020-12-06
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
领导文萃(2017年6期)2017-03-24
学与玩(2017年11期)2017-02-16
中学生数理化·高一版(2016年7期)2016-12-07
应用海洋学学报(2015年4期)2015-11-24
中学生数理化·中考版(2015年12期)2015-09-10
郑州大学学报(理学版)(2014年2期)2014-03-01
