
基于大数据挖掘与可视化的HPV病毒基因组分析研究
2020-10-27 11:27陈翠霞曹宗富李天君于磊喻浴飞蔡瑞琨罗敏娜李乾沈玥陆超高华方马旭
生殖医学杂志 2020年10期
陈翠霞,曹宗富,李天君,于磊,喻浴飞,蔡瑞琨,罗敏娜,李乾,沈玥,陆超,高华方,马旭*
(1.国家卫生健康委科学技术研究所,北京 100081;2.国家人类遗传资源中心,北京 102206)
宫颈癌是中国女性最常见的恶性肿瘤之一,仅次于乳腺癌[1]。宫颈癌主要病因是高危人乳头瘤病毒(Human papilloma virus,HPV)的持续性感染[2]。目前,宫颈癌治疗手段以毁损性手术为主,缺乏特效无创的阻断方法,临床检测鉴定方法仅覆盖23种亚型,占现存亚型10%,无法避免假阳性率和漏诊率问题,因此有必要对所有HPV进行快速准确的分型检测[3-6]。最新的疫苗只覆盖9种亚型的HPV病毒,保护71%的患者[3-6]。随着高通量测序技术的成熟,病原体的溯源、进化关系、基因组比较、致病危险性预测等研究工作已成为传染病的防控中重点研究方向[7-10]。有研究工作通过相同亚型HPV病毒的某段基因核苷酸的变异或基因整合热点位置,来研究病毒进化与疾病的相互关系以及病毒基因整合致病机制[10-13],也有文献对13种高危亚型的HPV病毒感染后其致病危险性发展的自然史进行监测研究[14]。但仍旧缺乏一种简单易操作的自动化流程来对HPV基因组大数据进行全面深入的挖掘分析。鉴于此,本文基于比较基因组学研究方法,针对HPV病毒全基因组数据,设计和集成一系列的算法工具包,构建了一种基于大数据挖掘技术的HPV基因组信息可视化分析流程框架,不仅覆盖迄今为止发现的298种亚型的HPV病毒,且具备对HPV病毒基因组数据的深度比对挖掘能力。
方 法
HPV基因组数据挖掘分析包含3个步骤(图1):(1)数据预处理:从搜集HPV基因组数据最全面的genBank(genBank[http://www.ncbi.nih.gov/nucleotide/SRA/genBank]数据库下载所有病毒类基因组数据作为原始数据。采用Perl 5.0语言脚本从原始数据中抽提298个HPV病毒亚型的全基因组数据,及其关键核心基因(E6、E7)的核酸和氨基酸序列,采用Perl脚本将关键基因按照染色体上的顺序连接组装,建立关键核心基因组,全部整理为fasta格式。(2)系统进化分析:采用Dnasp[15]软件评估全基因组序列的替换饱和度,对于通过评估筛选的HPV全基因组,采用MAFFT[16]软件进行多序列比对,并采用FastTree[17]软件的最大似然法完成进化树的构建,使用Dendroscope 3.0[18]软件实现全基因组进化树的展示、编辑和导出,至此完成全基因组进化树的构建。另外,关键核心基因组进化树的构建方法与全基因组进化树构建方法相同。(3)氨基酸分类比较分析:基于序列组分和分类比对进行氨基酸表达偏好模式分析。通过Perl脚本统计在HPV关键核心基因组的氨基酸序列中,20种氨基酸的占比,然后根据氨基酸的亲疏水性、极性、酸碱性分类,汇总为HPV病毒氨基酸分类的占比,用R语言的Scale包对结果进行归一化校准后得到氨基酸分类矩阵,最后用R语言的Heatmap包绘制氨基酸分类矩阵的热图(Heatmap),进而研究基因组分子结构和氨基酸表达偏好模式与毒株生物学特性表型的关系。

图1 HPV基因组数据挖掘分析框架流程图
结 果
核酸和氨基酸分子水平结构、氨基酸分类导致物种系统发育进化的不同,进而构成HPV致病危险性和主要感染侵袭部位不同的基础条件。所以本文从基于分子结构差异的系统进化分析和氨基酸表达偏好模式比较分析两个方面,来研究他们与亚型分化和致病危险性之间的关系。
1.数据预处理:抽提出上述298种HPV病毒的全基因组和核心关键基因(E6、E7)核酸和氨基酸序列,采用Perl脚本将关键核心基因按照染色体上的顺序连接组装,建立关键核心基因组,全基因组序列与关键核心基因组序列均为fasta格式。从这298种HPV中选出致病危险性较为明确的毒株37个和5个侵袭感染类型或致病危险性不太明确的毒株[1-6,10,12-14,19-34],共42个毒株,将其标记感染类型和危险性分类(字母S表示主要侵袭感染皮肤Skin;M表示主要侵袭感染粘膜Mucosa;N表示不明确;HR表示致病性为高危类型High-Risk;LR表示致病性为低危类型Low-Risk)后,与剩余的其他HPV病毒基因组作为参比毒株进入后续分析。统计得知参比的HPV全基因组长度范围为7 080~8 104 bp。


图2 42个HPV参比毒株在目前现存的298株HPV全基因组进化树上的分布情况
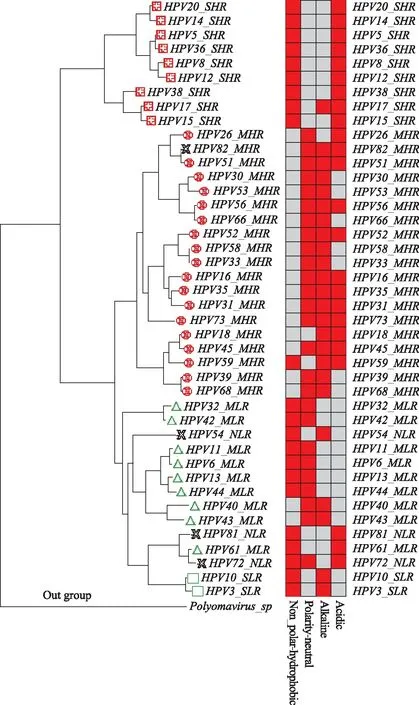
热图的红色块表示对该类型氨基酸较为偏好,占比较高;灰色块表示该类型氨基酸占比较少;热图第一列是 非极性/疏水性(Non_polar-hydrophobic)类型氨基酸,第二列是极性/中性(Polarity-neutral)类型氨基酸, 第三列是碱性(Alkaline)类型氨基酸,第四列是酸性(Acidic)类型氨基酸图3 42个参比HPV毒株的关键核心基因组进化树与其氨基酸分类热图对应关系图
3.氨基酸分类比较分析:基于进化树上毒株的位置,对42个参比毒株的关键核心基因组进行氨基酸序列组分和分类比对分析,进而发现氨基酸表达偏好模式与毒株生物学特性表型之间的关系(图3)。通过Perl脚本分别统计HPV关键核心基因组的氨基酸序列中20种氨基酸各自在整条序列中所占百分比,然后将氨基酸分为非极性/疏水性(Non_polar-hydrophobic)、极性/中性(Polarity-neutral)、碱性(Alkaline),酸性(Acidic)四大类,汇总HPV病毒20种氨基酸的百分比为四类氨基酸的占比,然后用R语言的scale包对结果进行归一化标准化后,得到氨基酸分类矩阵,最后用R语言的heatmap包绘制氨基酸分类矩阵的热图(Heatmap),从而可以展示氨基酸表达偏好模式的差异与毒株生物学特性表型的关系。由图3可见,皮肤高危型(SHR,)毒株比较偏好表达非极性/疏水性和酸性氨基酸产物;黏膜高危型(MHR,)毒株比较偏好表达极性/中性和碱性氨基酸产物,其中毒性较高的HPV16,HPV31,HPV18,HPV35,HPV73,HPV56[14]毒株也同时偏好表达酸性氨基酸;黏膜低危型(MLR,)毒株比较偏好表达非极性/疏水性和极性/中性氨基酸产物,有趣的是HPV40_MLR和HPV43_MLR的氨基酸表达种类模式与MHR的模式很类似,也偏好极性/中性和碱性氨基酸产物,HPV61_MLR和HPV81_NLR的表达模式与SHR类似,也偏好表达非极性/疏水性和酸性氨基酸产物,这也许正是HPV各亚型之间转化以及低危和高危类型之间转化的分子水平的物质基础;皮肤低危型()比较偏好表达非极性/疏水性和碱性氨基酸产物。
讨 论
HPV病毒的亲缘性或危险性与进化树上位置密切相关,而进化树是基于基因组分子组分和结构之间的遗传距离绘制的。由图2、图3可见,HPV病毒根据亲缘关系远近汇聚到不同的簇,聚集位置相近,说明可能是由同一祖先共同进化而来,致病危险性也相似。如果毒株呈现独立分支,可以预测该毒株是新亚型。本方法曾成功应用于链球菌新菌的分离鉴定工作[36-37],而本文将该方法推广应用到HPV基因组分析,结果显示全基因组进化树(参见图2)的Clade分布趋势与关键核心基因组进化树(参见图3)结果相吻合,一方面表明关键核心基因E6/E7主导了HPV物种的进化和分化,从分子进化的角度验证E6/E7蛋白对宫颈癌的发生发展起决定性的作用。同时证明了本流程框架从细菌基因组分型研究[36-37]推广应用于病毒分型研究仍然是可行的,可以辅助HPV病毒新亚型鉴定、亚型间亲缘性和危险性的预测,为HPV病毒的防控提供依据。例如HPV82_NHR,已知属于高危亚型,但是侵袭部位不很明确,进化树上与粘膜高危型聚集到一个clade(参见图2、图3左),且其氨基酸偏好表达模式与MHR相同(参见图3右),因此预测其侵袭感染部位为粘膜类型,即为粘膜高危型(HPV82_MHR);HPV8_SNR,已知其属于侵袭皮肤类型,进化树上与皮肤高危型聚集到一个clade(参见图2、图3左),且其氨基酸偏好表达模式与SHR相同(参见图3右),因此可以预测其为皮肤高危型(HPV8_SHR);同样,HPV54_NLR、HPV72_NLR均与粘膜低危型聚集到一个clade(参见图2、图3左),同时两个亚型毒株的氨基酸偏好表达模式与MLR相似,可以预测他们为粘膜低危型,即分别为HPV54_MLR、HPV72_MLR。因此可以说,图2、图3从分子进化和氨基酸分类偏好模式角度验证了文献[19-22]结果的正确性,致病危险性类似的毒株,其基因组结构特点也类似,找到了HPV病毒的生物学表型特性的差异与核酸、氨基酸分子水平差异有紧密相关性的证据。
基于不同的氨基酸类型在基因组序列中的表达比率,来研究HPV基因组中氨基酸的表达偏好模式,进而发现氨基酸表达偏好模式与病毒侵袭皮肤的特性之间的关系。由图3可见,致病危险性相似的毒株,氨基酸表达偏好模式也类似,从而保证了其物种基本的生物学特性。MHR和MLR共同偏好表达极性/中性(Polarity-neutral)类型氨基酸,可能说明极性/中性氨基酸的偏好表达与病毒侵袭粘膜的特性有密切关系,而MHR因为更加偏好表达碱性和酸性氨基酸而成为高危亚型,MLR因为更加偏好表达非极性/疏水性氨基酸产物而成为低危亚型;SLR和SHR共同偏好非极性/疏水性,可能表明非极性/疏水性氨基酸的高表达与病毒侵袭皮肤的特性有密切关系,SHR因为更偏好表达酸性氨基酸而成为高危亚型,SLR因为更偏好表达碱性氨基酸而成为低危亚型。基因组核酸分子结构和基因结构的差异,氨基酸偏好表达模式的不同,均导致了HPV病毒侵袭特性的不同和致病危险性的多样化。
综上所述,本研究具有以下特点:(1)目前研究最多的HPV亚型数量是131个,本文囊括目前发现的298种HPV亚型毒株代表株,并整理获得HPV全基因组序列和病毒所有基因(E1/E2/E4/E5/E6/E7/L1/L2)的序列、起止位点、长度、分离病毒的亚型和名称等详细信息。(2)通过全基因组比较和系统进化关系分析,可以根据新发病毒在已知致病危险性毒株的进化树上的分布情况,实现对未知或新发病毒的亚型、感染侵袭特性及其致病危险性的快速预测,为病毒的预防和控制提供证据支持和技术补充。(3)本文也尝试探究氨基酸表达偏好模式与致病危险性之间的关系,流程框架能够迅速发现其氨基酸表达偏好模式,利用该表达模式推断HPV毒株的侵袭特性和致病性危险性,尝试将基因组结构差异和氨基酸偏好表达模式与生物表型特性做关联性研究,为指导临床用药和控制感染提供依据。
猜你喜欢
天津市教科院学报(2021年5期)2021-11-10
生物学通报(2021年9期)2021-07-01
中华养生保健(2020年7期)2020-11-16
时代英语·高一(2019年5期)2019-09-03
天然产物研究与开发(2018年9期)2018-10-08
健康必读·下旬刊(2018年4期)2018-06-04
中国医学创新(2017年7期)2017-03-31
江苏农业科学(2016年8期)2017-02-15
天然产物研究与开发(2016年1期)2016-06-05
中国卫生标准管理(2015年25期)2016-01-14
