
Ni2Bn(n= 1~10)团簇的结构与稳定性研究
2020-10-21 07:49:12郝爱琴薛海波
山西师范大学学报(自然科学版) 2020年3期
郝爱琴,薛海波
1.山西师范大学分析测试中心,山西 临汾 0410002.山西师范大学化学与材料科学学院,山西 临汾 041000


1 计算方法
所有团簇的结构优化都采用VASP软件包,基于密度泛函理论(DFT)方法完成.体系的交换相关能采用自旋极化的广义梯度近似(GGA)的Perdew Burke Ernzerhof(PBE)泛函来描述.价层电子采用平面波基组来描述,平面波函数截止能设为600 eV.电子-离子实相互作用采用缀加平面波(PAW)方法描述.对于B原子,2s和2p电子被视为价层电子,对于Ni原子,3d和3s电子被视为价层电子.Ni2Bn团簇被放置在一个15 Å×15 Å×15 Å的立方体超晶胞中,两个相邻团簇之间的相互作用可以忽略不计.结构优化时,没有对称性限制.k点网格的大小设置为1×1×1.结构优化时,力的收敛标准为0.01 eV/Å,且总能量的变化小于10-5eV.频率计算以确保得到的团簇在处在势能面上的极小值点,同时获得了零点振动能(ZPE).
为了确定Ni2Bn(n=1~10)团簇的最稳定结构,我们充分考虑了各种可能的初始构型:包括线型、平面型和三维立体型结构.对于小团簇(n≤5),除了独立设计团簇结构以外,我们还将之前计算的稳定纯Bn+2簇中的B原子替换为Ni原子,或者将Ni原子放在稳定的纯Bn簇中从而考虑各种可能存在的合理的初始结构.对于n≥6的Ni2Bn团簇,我们利用CALYPSO结构搜索软件基于粒子群优化(PSO)算法进行了全局搜索,以保证我们找到能量最低的Ni2Bn团簇.
2 结果与讨论
2.1 几何结构
采用如上所述的团簇构建方法及结构优化方法,我们得到了Ni2Bn(n=1~10)团簇的最稳定结构,如图1所示.图1中我们还列出了相应团簇的次稳定结构和它们的相对能量、自旋磁矩和对称性.

图1 Ni2Bn(n=1~10)团簇的最稳定与次稳定异构体及其相对能量、磁矩和对称性Fig.1 The most stable and the second stable isomers of Ni2Bn(n=1~10) clusters,with their relative energies,magnetic moments and symmetries
Ni2B团簇的最稳定结构(1a)是一个以B原子为中心的线性结构,B—Ni距离为1.75 Å.次稳定结构(1b)是一个等腰三角形,顶角为50.5°.两个相等的B—Ni距离为1.78 Å.(1b)的能量比(1a)高0.17 eV.Ni2B2团簇的最稳定结构为对称性呈D2h的菱形(2a),B—Ni键长为1.81 Å,比Ni2B中的键(1.78 Å)要长.B—B键长为1.88 Å,明显长于在相同理论水平下B2(1.61 Å)中B—B键长.次稳定的Ni2B2异构体(2b)是对称性为C2v的扭曲菱形,其能量比(2a)高0.93 eV.对于Ni2B3,我们考虑了所有可能的线性结构,平面和三维(3D)共11个异构体.三角双锥体(3a)被确定为最稳定结构.在(3a)中,B—Ni键的长度约为1.88 Å,还观察到非常长的B—B键,长度约1.79 Å.梯形平面异构体(3b)为次稳结构,其能量比(3a)高0.10 eV.最稳定线性结构的能量比(3a)高2.58 eV(未经ZPE校正),这表明较大Ni2Bn团簇的线性结构并不稳定.对于Ni2B4,我们不需要考虑线性异构体,只优化了所有平面结构和3D异构体.Ni2B4的最稳定异构体具有平面扇形结构(4a),其B—B键长为1.54 Å~1.77 Å,比Ni2B3中的B—B之间的键长要短,B—Ni键长为1.80 Å~1.91 Å.值得注意的是,M2B4(M=Sc,Ti,V和Ta)的最稳定结构均为不完整的双锥体.Fe2B4和Co2B4也均具为3D结构.次稳定的Ni2B4也具有平面结构,为一平行四边形(4b),其能量比(4a)高0.27eV.Ni2B5的基态结构和次稳定结构均为平面结构,对称性均为C2V.两个异构体能量仅差0.02 eV.
Ni2Bn(n=2~6)的结构演变明显不同于先前报道的团簇V2Bn、Ti2Bn、Sc2Bn和Ta2Bn、V2Bn、Ti2Bn、Sc2Bn和Ta2Bn更趋于形成不完整的双锥体.Ni2Bn(n≤6)团簇,明显表现出立体结构与平面结构的竞争.如图所示,除了Ni2B3的最稳定结构为三角双锥以外,Ni2B4和Ni2B5的最稳定结构与次稳定结构都是平面或准平面结构.Ni2B6的次稳定结构也呈平面结构.当n≥7时,最稳定结构都不是理想的双锥结构,Ni2Bn的这种结构变化趋势主要与两个因素有关:一个是Ni原子半径要小于Sc、Ti、V等,不易形成6以上的双锥结构.二是Ni原子d轨道上电子较多,在成键性上不同于Sc、Ti、V等前过渡金属原子,在小团簇时,更易形成平面结构.
2.2 相对稳定性
我们还计算了平均结合能来研究团簇的相对稳定性.平均结合能(Eb)是衡量簇合物体系相对稳定性非常重要的指标之一,其定义如下:
Eb(Ni2Bn)=[nE(B)+2E(Ni)-E(Ni2Bn)]/(n+2)
(1)
其中,E(B),E(Ni)分别是一个B原子和一个Ni原子的能量.E(Ni2Bn)是团簇Ni2Bn最稳定结构的能量.在图2中可以看出,随着团簇尺寸的增大,平均结合能随着硼原子数量的增加而呈递增趋势.从n=1~7,平均结合能Eb值迅速增大,然后,从n=8~10,Eb值变化缓慢.在n=7处出现局域最大值,这说明Ni2B7团簇具有更高的稳定性.

图2 基态Ni2Bn(n=1~10)团簇的平均结合能Fig.2 Average binding energy of ground state Ni2Bn(n=1~10) clusters
为了深入研究Ni2Bn(n=1~10)团簇尺寸对团簇相对稳定性的影响,我们计算了团簇Ni2Bn的二阶差分能量值Δ2E,其定义为:
2Ni2Bn→Ni2Bn+1+ Ni2Bn-1
Δ2E=E(Ni2Bn+1)+E(Ni2Bn-1)-2E(Ni2Bn)
(2)
其中,E(Ni2Bn)是指经过弛豫后Ni2Bn团簇最稳定结构的能量.Δ2E可以反映相邻团簇之间的相对稳定性.为了更全面地衡量团簇Ni2Bn的相对稳定性,我们还定义了另一个差分能量Δ2E(2),如下公式所示:
2Ni2Bn→Ni2Bn+2+ Ni2Bn-2
Δ2E(2)=E(Ni2Bn+2)+E(Ni2Bn-2)-2E(Ni2Bn)
(3)
Ni2Bn团簇的Δ2E和Δ2E(2)与团簇尺寸n之间的关系如图3所示.可以发现Ni2Bn团簇的Δ2E呈不规则振荡,与V2Bn和Ti2Bn团簇的奇偶振荡变化明显不同.Δ2E揭示Ni2Bn(n=2,4,7)的团簇比其相邻团簇更稳定.但Δ2E(2)揭示Ni2B3和Ni2B7更稳定.综合Δ2E和Δ2E(2)我们得出的结论是Ni2B7是幻数团簇,这也与前边结合能变化规律一致.我们以前的研究表明,Sc2Bn的幻数为n=7和8;Ti2Bn的幻数为n=6,7,8;V2B6是V2Bn的幻数团簇;Fe2Bn的幻数为n=6和7;Co2B10是Co2Bn的幻数团簇.但它们的幻数团簇都为完美的双锥结构.

图3 基态Ni2Bn(n=1~10)团簇的二阶差分能量值Fig.3 SecondorderdifferentialenergyvalueofgroundstateNi2Bn(n=1~10)图4 Ni2Bn(n=1~10)团簇基态结构的垂直电离势(VIP)、垂直电子亲和能(VEA)和化学硬度(η)Fig.4 Verticalionizationpotential(VIP),verticalelectronaffinity(VEA)andelectronhardness(η)ofthegroundstatestructureofNi2Bn(n=1~10)clusters
2.3 电子性质
为了分析团簇Ni2Bn的尺寸和电子稳定性的依赖关系,我们计算了团簇Ni2Bn的垂直电离势(VIP)和垂直电子亲和能(VEA).VIP和VEA的定义如下:
(4)
(5)
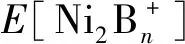
3 结论
采用电子密度泛函理论方法下的广义梯度近似对Ni2Bn(n=1~10)团簇的几何形状、稳定性、电子结构做了系统研究.研究结果表明Ni2Bn(n=2~6)的结构演变明显不同于先前报道的V2Bn、Ti2Bn、Sc2Bn和Ta2Bn团簇.V2Bn、Ti2Bn、Sc2Bn和Ta2Bn更趋于形成不完全的双锥体.而Ni2Bn(n≤6)团簇明显表现出立体结构与平面结构的竞争.Ni2B4和Ni2B5的最稳定结构与次稳定结构都是平面或准平面结构.Ni2B6的次稳定结构也呈平面结构.当n≥7时,最稳定结构倾向于3D结构,但都不是理想的双锥结构.通过分析团簇的平均结合能、二次差分能量及VIP、VEA和化学硬度,我们得出Ni2Bn的幻数团簇为Ni2B7,其不仅具有较高的热力学稳定性,也具有较高的动力学稳定性.除Ni2B6外的自旋磁矩为2 μB外,Ni2Bn的自旋磁矩对于偶数n时为0 μB,对于奇数n为1 μB.
猜你喜欢
吉林大学学报(理学版)(2025年1期)2025-02-06 00:00:00
系统仿真技术(2022年4期)2023-01-17 13:01:44
大学物理(2022年9期)2022-09-28 01:10:52
云南化工(2021年8期)2021-12-21 06:37:38
物理通报(2020年7期)2020-07-01 09:28:02
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
信息记录材料(2016年4期)2016-03-11 15:22:30
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
