
促红细胞生成素对神经退行性疾病的作用
2020-10-20 03:04许恒正吴志猛程孝中
生物学杂志 2020年5期
许恒正, 吴志猛, 程孝中
(1. 滁州城市职业学院 基础部, 滁州 239000; 2. 江南大学 教育部糖化学与生物技术重点实验室,无锡 214122; 3. 合肥师范学院 生命科学学院, 合肥 230601)
迄今为止,神经退行性疾病还缺乏有效的疗法。针对每种特定神经退行性疾病的一线疗法尚在研究之中,如表观遗传疗法、基因疗法、干细胞疗法,甚至3-D类器官等。但近几年来,单一药物疗法的EPO作为治疗剂在神经退行性疾病中的作用成为研究热点。
促红细胞生成素(Erythropoietin, EPO) 化学本质为糖蛋白,属于一类细胞因子,其主要功能为促进骨髓中的红系祖细胞增殖、分化为成熟的红细胞[1]。近几年来,越来越多的证据显示,EPO在血管生成、炎症反应、氧化应激、细胞凋亡等方面发挥了重要作用,这些作用表明EPO具有潜在的保护作用,如对心血管、肾脏、干细胞及神经系统方面的保护作用[2]。
1 EPO的分子结构及神经保护作用机理
EPO基因位于第7号染色体上,编码前体的EPO蛋白由193个氨基酸组成,其中27个氨基酸残基作为前导分泌肽在形成成熟的EPO时被切除,C末端的精氨酸也除去,最终形成165个氨基酸构成的成熟糖蛋白EPO,相对分子质量为 30.4 ku[3]。分子中共有4个糖基化位点,其中3个为N-连接糖基化位点(Asn24,Asn38,Asn83),1个为O-连接糖基化(Ser126)位点。EPO中第33位和29位、第161位和7位的半胱氨酸侧链巯基形成2对二硫键,最终EPO呈α螺旋构象(图1)[4]。
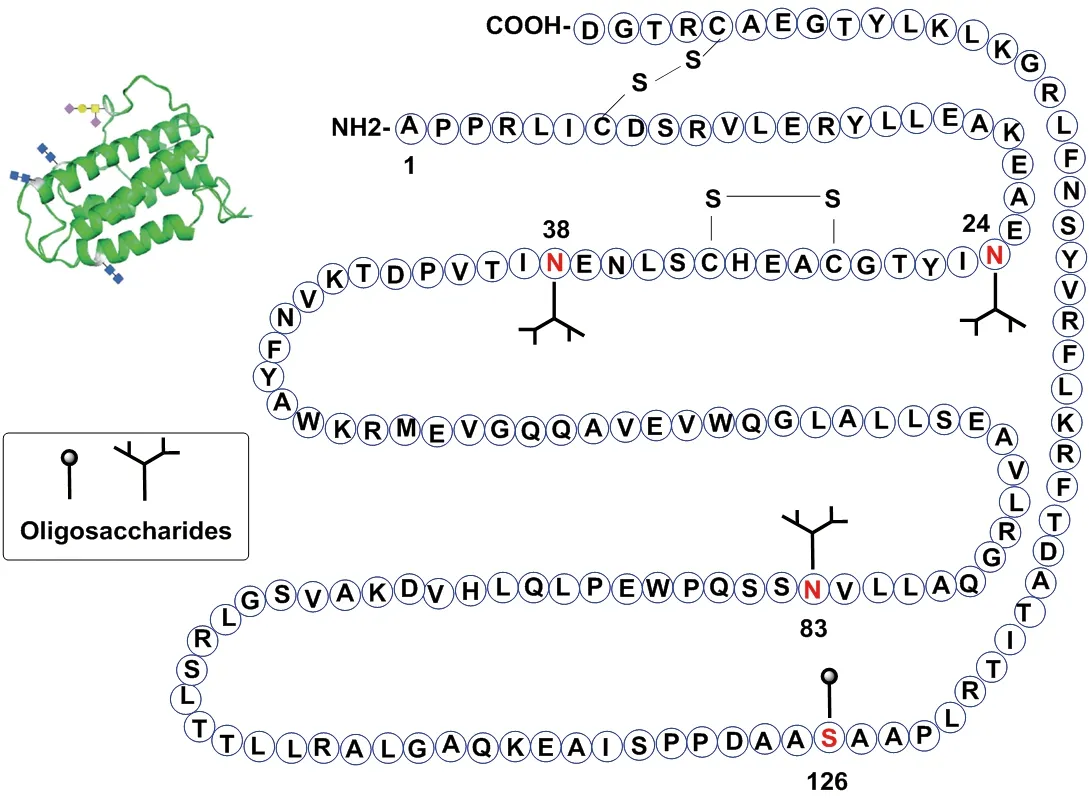
图1 EPO的分子结构
EPO通过与促红细胞生成素受体(Erythropoietin receptor, EPOR)结合发挥神经保护功能。EPOR属于整合膜蛋白,以单体形式存在,包括由225个氨基酸组成的胞外结构域、23个氨基酸构成的跨膜结构域和235个氨基酸组成的胞内结构域[5],胞外结构域包括D1和D2两个EPO结合结构域。EPO与EPOR结合后,引起EPOR构象发生改变,EPOR同源二聚化从而引起胞内结构域上结合的JAK2酪氨酸激酶被活化,活化的JAK2激酶磷酸化下游激酶,从而引起多个下游信号通路活化(图2-A)[6]。这些信号通路主要有转录因子5(STAT5) 、促分裂原活化蛋白激酶(MAPK)、磷脂酰肌醇3-激酶(PI3K/Akt)等[7]。EPO还可以通过EPOR单体作用方式进行信号转导(图2-B)。
2 EPO对神经退行性疾病的作用
2.1 帕金森病
帕金森病(Parkinson′s disease,PD)是第二大最常见的神经退行性疾病。它的症状主要是运动迟缓,震颤和肢体僵硬[8]。两个主要的病理标志是多巴胺能神经元的凋亡和α突触核蛋白聚集,这些聚集加剧神经细胞的氧化应力和炎症反应,最终导致细胞毒性[9]。研究发现,在许多体内或体外的PD模型中,EPO均表现出神经保护作用。例如,被EPO与1-甲基-4-苯基吡啶鎓共同处理的交感神经细胞PC12表现出良好的生存状态,而PC12细胞经6-羟基多巴胺预处理,再给予EPO刺激,PC12细胞的生存状态同样得到改善。EPO的神经保护作用主要是通过抑制α突触核蛋白聚集导致的神经细胞氧化、炎症反应和细胞凋亡来实现[10]。
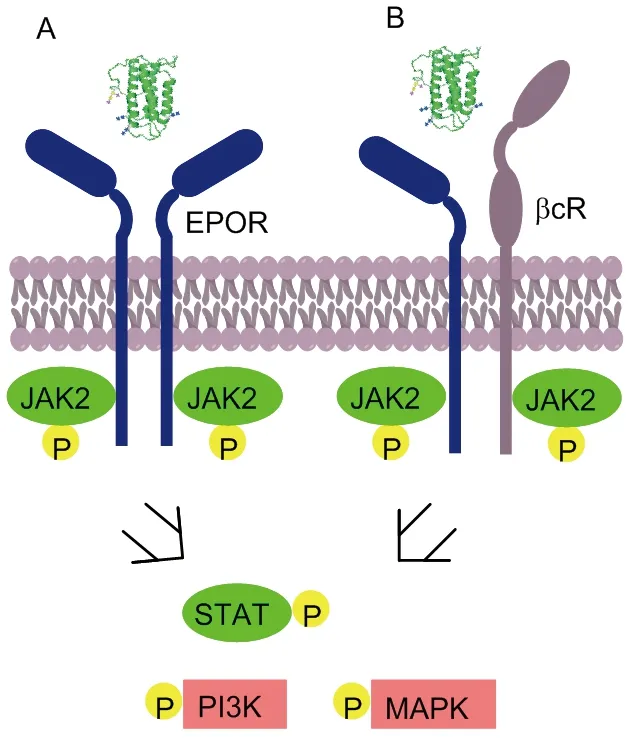
A: EPOR同源二聚化;B: EPOR单体与βcR受体相互作用
EPO可以抑制不同神经细胞(星形胶质细胞、小胶质细胞和神经元细胞)的炎症反应,从而抑制神经退行性变化[11]。例如,炎症反应所释放的炎症因子往往导致神经细胞肿大,在星形胶质细胞的损伤模型中,一种依赖于MAPK通路的水孔蛋白AQP4会大量表达,而重组的EPO可以下调AQP4表达,从而缓解细胞肿大,其主要依赖的信号通路则为MAPK途径,但EPO下调AQP4表达的具体机制尚不清楚[12]。而在神经胶质细胞损伤模型中, EPO则是通过降低中间丝蛋白表达量从而抑制神经胶质细胞肥大,降低神经细胞的炎症反应[13]。这说明在不同类型神经细胞中,EPO的神经保护作用机理也是不一样的。另外一种EPO调节神经炎症的机制则是通过TNF-α实现,在鱼藤酮和6-羟基多巴胺诱导的PD动物模型中,EPO显著降低TNF-α的水平,恢复了PD大鼠体内酪氨酸羟化酶(TH)水平,此处EPO通过活化Akt信号,调节NF-κB p65亚基降低炎症因子TNF-α的表达,从而实施神经保护作用[14-15](图3)。另外,内源性的EPO(如由神经前体细胞分泌的EPO)同样能够缓解早期炎症,促进PD小鼠模型中的神经元的存活,并能恢复TH水平[16-17]。
神经元细胞凋亡是PD一个重要的病理特征,在6-羟基多巴胺诱导的PD细胞模型中,EPO可以抑制6-羟基多巴胺诱导的多巴胺神经元毒害作用。EPO与EPOR结合后,通过磷酸肌醇-3激酶途径(PI3K)活化Akt,抑制下游凋亡相关分子caspase 9/3表达和核酸内切酶活性,从而抑制DNA断裂,最终达到抗凋亡作用(图3)。此外,EPO也可以经过ERK1/2途径实现抗凋亡作用,但该途径在PD模型中并不常见[18-19]。
由此可见,EPO在PD模型中的神经保护主要通过EPO/EPOR下游不同信号通路来下调神经炎症反应和细胞凋亡相关分子,以达到缓解神经细胞因α突触核蛋白聚集所导致的氧化应激目的,而氧化应激可以认为是导致神经细胞炎症反应和细胞凋亡上游事件。另外,此处涉及到的炎症反应和细胞凋亡通路也是经典的作用途径,EPO是否可以影响其他非经典途径并不清楚,这也为我们继续探索EPO在PD中的保护机制提供了一个新的视角。
2.2 阿尔茨海默氏病
阿尔茨海默氏病(AD)即为常见的老人痴呆症,临床特征为逐渐丧失记忆和认知功能下降。到目前为止,尚未找到AD的治愈方法,因此,评估诸如EPO之类的新方法和新分子的治疗潜力具有重要意义。多项研究显示EPO在AD治疗方面表现较好,如在细胞水平上,EPO在PC12细胞和原代神经元细胞中能够抑制细胞凋亡和减轻Aβ聚集导致的细胞毒性作用[20]。AD疾病在细胞水平上的重要标志是存在于细胞外的淀粉样蛋白(Aβ)大量聚集,影响了神经细胞膜的稳定性,导致阳离子大量进入细胞,诱导细胞凋亡,EPO主要通过阻止凋亡途径相关分子的活化,实现神经细胞保护[21]。
被EPO活化后的EPOR激活JAK / STAT,并磷酸化下游RAS和PI3K途径。其中的RAS途径的激活可以促进细胞核中Bcl家族蛋白大量表达(如Bcl2和Bcl-xL),Bcl2和Bcl-xL可以阻止凋亡蛋白半胱氨酸激酶caspase的活性[22-23](图3)。EPO这种作用已通过siRNA技术在星形胶质细胞中得到证明,另外,使用PC12细胞的体外实验也证明了EPO对Aβ诱导的毒性的神经保护作用[24]。在Aβ25-35诱导的AD细胞模型中,EPO是通过STAT下游的另一条途径PI3K/AKT实现神经保护,EPO通过PI3K/AKT途径抑制糖原合酶激酶3β(GSK-3β)的磷酸化。正常情况下,活化的GSK-3β可以促进凋亡蛋白caspase释放[24-25](图3)。
在小胶质细胞系EOC2模型中,EPO抗凋亡作用则与Wnt1和PI3K介导的途径相关[26]。Wnt1通过维持线粒体膜电位,促进Bad从线粒体到细胞质转运,减少Bad/Bcl-xL复合物的形成并增加Bcl-xL/Bax复合物来调节细胞凋亡级联反应,阻止caspase 1和caspase 3的激活[3](图3)。在小胶质细胞中施用EPO(10 ng / mL)可以显著维持Wnt1的表达,这可能是防止Aβ诱导的神经毒性的另一种保护机制[27]。
AD与PD在病理特征上具有一定的相似性,在AD中,神经细胞因胞外淀粉样蛋白大量聚集而导致神经细胞氧化应激、炎症反应和细胞凋亡。因此,EPO在AD中的保护机制和在PD中特别类似,主要也是通过EPO/EPOR下游不同信号途径来抑制炎症和细胞凋亡,例如,通过磷酸肌醇-3激酶途径(PI3K)活化Akt,抑制下游凋亡相关分子caspase。
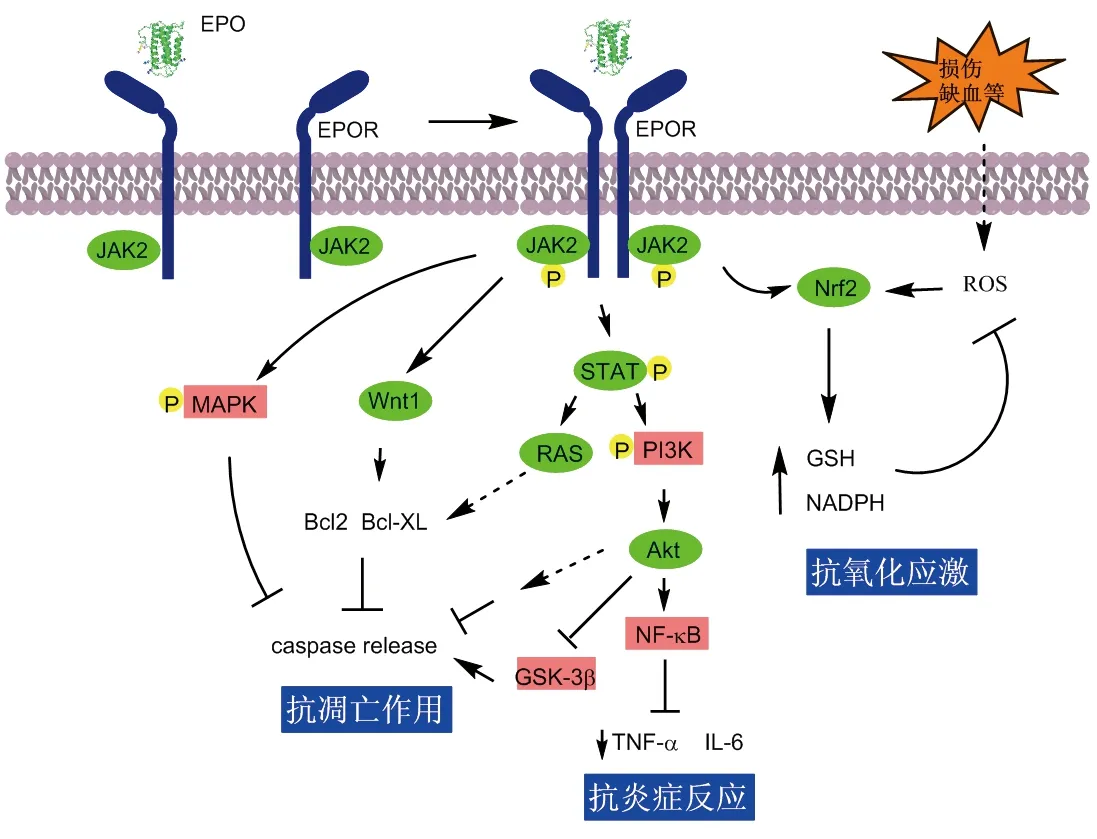
图3 EPO神经保护作用机制有关通路
2.3 脊髓损伤
脊髓损伤(Spinal cord injury,SCI)是由高强度创伤所导致,其特征是严重的神经功能从感觉丧失到部分或完全肢体麻痹。创伤释放有毒物质,如自由基、脂质过氧化物酶、类花生酸和谷氨酸,最终导致神经元和神经胶质细胞凋亡[28]。在体外和体内都证明了EPO的神经保护作用,其保护机制主要有抗凋亡、抗氧化和抗炎症作用[29]。
脊髓损伤主要是由物理性创伤所导致,会释放大量的氧自由基,引起细胞氧化应激反应。EPO缓解SCI的机制之一就是抑制细胞氧化毒性作用,核因子Nrf2信号通路是目前发现的抵御外源性刺激物的抗氧化应答反应的核心转录因子,在炎症和氧化应激下,Nrf2从细胞质入核调控细胞保护酶(如NADPH、GST)大量表达,从而缓解细胞氧化和炎症,外源给予重组EPO可以激活Nrf2信号通路[30](图3)。
2.4 脑缺血
缺血性中风通常是由于脑动脉血栓或局部血栓形成的闭塞引起的,从而引起急性缺血性中风。缺血性损伤可以使ATP减少,Na+/K+泵和ATPase遭到破坏,从而导致严重的离子动态失衡。后期一氧化氮的释放、质膜的破坏和促炎性细胞因子释放均会使神经兴奋性毒性增强,最终导致凋亡分子(caspase)激活。因此,caspase激活,特别是在神经元凋亡过程中过表达的caspase 3可能是缺血性损伤导致细胞死亡的主要原因[31]。
正常情况下,脑中EPO和EPOR的表达极少,但是在诸如缺血的损伤情况下,在内皮细胞、星形胶质细胞和神经元内HIF被激活,导致下游分子EPO表达上调[32]。如在经历脑缺血的海马和皮质神经元中, EPO预处理大脑皮层细胞能够激活抗凋亡基因(如XIAP和c-IAP2),表明EPO参与抗凋亡和细胞恢复作用[33]。
与其他神经退行病变类似,EPO在脑缺血中的保护机制也涉及到抗炎症作用。在兔的模型中,重组的EPO在大脑缺血期间具有神经保护作用,同时坏死皮质神经元明显减少[34],EPO募集白细胞和促炎细胞因子(IL-6和TNF),这表明EPO可以减轻局部缺血引起的炎症反应。
脑缺血和脊髓损伤都是由外在突发的、有一定物理强度的创伤导致的神经病变,引起的细胞损伤主要还是细胞膜损伤、氧化应激为主导的事件。因此,相对于慢性进展的AD和PD来说,EPO在SCI和脑出血模型中通过经典的Nrf2信号通路来抑制氧化应激反应,以及非经典的抗凋亡基因XIAP和c-IAP2来抑制细胞凋亡。
3 结论与展望
EPO作为一种安全用药在临床已经应用了20多年,主要用于肾脏疾病、肿瘤等导致的恶性贫血治疗。最近几十年,大量动物试验证实,EPO具有神经保护作用,并得到广泛认可,但在临床研究中,EPO的治疗效果并不完全受到肯定,其在临床神经疾病方面的治疗仍然存在一些争议,如静脉注射EPO 对弥漫性轴索损伤的患者进行治疗时,血液中红细胞数量并未改善。因此,对EPO在神经保护机制的研究有待进一步加强,为EPO在临床神经疾病方面的治疗提供理论支撑。
EPO另一个发展方向为EPO衍生物在神经疾病方面的治疗作用,如氨甲酰化EPO(EPO部分氨基酸被氨甲酰化修饰),它与EPO的作用机制不同,选择性地与βCR 异源二聚体结合发挥神经保护功能。此外,EPO本身的糖基化位点也是发展EPO衍生物的关键部位。因此,EPO衍生物通过不同的作用机制可以克服EPO在治疗神经疾病方面的一些缺陷,提高治疗效果,其作用机制和药物活性方面是将来值得探索的领域。
猜你喜欢
昆明医科大学学报(2021年10期)2021-12-02
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
昆明医科大学学报(2021年2期)2021-03-29
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
神经损伤与功能重建(2018年2期)2018-02-01
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国学术期刊文摘(2016年8期)2016-02-13
中国医药生物技术(2015年4期)2015-12-26
