
卵巢癌基因共表达网络及预后标志物的研究
2020-10-20 02:25顾云婧
生物学杂志 2020年5期
顾云婧, 朱 平
(江南大学 理学院, 无锡 214122)
卵巢癌是最致命的女性疾病之一,2012年全球共诊断出约238 700例新增卵巢癌患者,居于女性癌症死因的第4位[1]。由于潜在症状不明显,且缺乏可靠的早期筛查方法,约70%卵巢癌患者被诊断时已为晚期(III-IV期),晚期患者的5年生存率低于30%[2]。目前,卵巢癌患者的治疗主要通过手术切除和化疗[3],但大多数晚期患者会在18个月内复发[4],预后情况并不理想。因此,这就需要对卵巢癌的早期诊断和预后标志物进行更深入的研究,将其应用于卵巢癌的治疗指导和患者管理,以开发更有效的治疗方法来改善卵巢癌的预后效果。
近30年,基因、蛋白质、代谢等组学的迅速发展以及对肿瘤生物学的深入研究,目前已经发现了VEGF、HE4等数百种具有预后价值的卵巢癌生物标志物[5]。因此,临床上迫切需要对卵巢癌预后标志物进行系统分析,以便于指导临床治疗策略,基因相互作用网络则为系统分析提供了可能。研究表明,基因相互作用网络可以用于探究功能基因簇(功能模块)和预后因素之间可能存在的内在联系[6]。通过加权基因共表达网络分析方法(Weighted gene co-expression network analysis,WGCNA)构建稳健的基因共表达网络,其中有意义的模块可用于推断肿瘤机制,预测患者存活率,以及建立新的诊断或治疗目标。WGCNA方法为系统生物学提供了功能性解释工具,目前已运用于乳腺癌、子宫内膜癌等肿瘤中[7-10]。在卵巢癌中,研究人员应用WGCNA研究了TP53错义和无效突变[11]。
为了提高预后标志物的准确性,在构建共表达网络时融合基因的蛋白质相互作用(Protein-protein interaction, PPI)数据,从而将基因的生物学意义纳入网络。此外,普遍认为拓扑上重要的(例如高度连接的)基因在疾病的发展中具有重要功能,并且倾向于在患者中表现出一致的表达变异[12]。因此,选择拓扑重要基因作为预测因子构建预后模型,能够使得模型的性能在以下两个方面得到改善:增强预后模型的稳健性,因为拓扑上重要的基因具有一致的表达变化,从而降低了结果的偶然性;提高所鉴定的基因与给定疾病的生物学相关性。
本文运用筛选得到的预后相关基因构建了卵巢癌预后加权基因共表达网络,并对网络进行系统分析。对于网络中的卵巢癌预后模块,整合模块中基因的PPI数据对模块网络进行重新加权,得到预后模块重加权共表达网络(Reweighted co-expression network for module prognosis-related genes, RMPCEN),并利用RMPCEN中基因的拓扑性质对模块基因进行排序。在同时考虑模型预测能力和模型基因个数的条件下,选择合适数量的生物标志物构建最终预后模型,预测卵巢癌患者的预后情况。
1 材料和方法
1.1 数据获取
卵巢癌患者的研究数据从癌症基因组数据库(The cancer genome atlas, TCGA)(https://portal.gdc.cancer.gov/)中下载,TCGA数据库收录了众多癌症的实验数据和临床数据。本文从数据库中下载了卵巢癌患者的全基因高通量转录组测序数据和临床信息,并对编码基因表达量进行归一化处理。最终选取了320个同时具有基因表达数据和临床信息的卵巢癌样本,且生存时间均大于30 d。
1.2 筛选预后相关基因
对于患者的临床数据,由于每个病例随访时间不同,发生终点事件的可能性也不相等。因此,本文选用Cox单因素回归分析[13]筛选与总体生存(Overall survival, OS)相关的基因,作为卵巢癌预后相关基因。对于每个基因,根据其表达值进行中值分割,将患者分为高表达和低表达两组进行单因素回归分析。回归分析采用R软件进行,P<0.05认为有统计学意义。
1.3 预后基因共表达网络的构建方法
本文采用加权的基因共表达网络分析方法构建卵巢癌预后网络,并对网络进行模块挖掘。
运用皮尔逊相关系数衡量基因之间的相关性,得到基因表达的相关性矩阵:
S=[Smn=|cor(m,n)|]
(1)
其中,Smn表示基因m和n的相关性系数。
选择适当的加权系数β作为软阈值,运用幂指数函数计算基因之间的邻接系数,将相关矩阵转换为邻接矩阵,使得网络节点之间的连接符合无尺度网络分布,即符合幂律分布,邻接矩阵:
A=[amn=power(smn,β)]=[|smn|β]
(2)
其中,amn表示基因m和n的邻接系数。
基于选定的软阈值,进一步地将邻接矩阵转化为拓扑重叠矩阵(Topological overlap matrix, TOM)[14]:
(3)
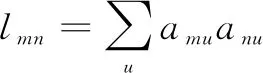
根据计算得到的基因拓扑重叠矩阵,运用层次聚类对网络进行划分,并识别网络中的模块。同时,运用Benjamini-Hochberg法校正模块中基因对相关系数P值,选择P<0.01的基因对构建模块网络。
1.4 模块网络的重加权及网络分析方法
为了提高预测效率,本文考虑模块网络的生物学意义,构建模块网络时通过融合基因的PPI数据对网络进行重新加权。从STRING[15]数据库(https://string-db.org)中下载了模块基因的蛋白质相互作用数据,利用PPI的置信度对网络重新加权:
(4)
对于重加权的模块网络进行拓扑特征分析,验证其是否属于小世界网络。网络特征分析包含网络的特征路径长度(Characteristic path length, CPL)、聚类系数(Clustering coefficient, CC)和小世界指数(Small-world index, SW)。网络的CPL为网络中所有节点对之间最短路径长度的平均值,节点的CC为该节点直接连接的节点中实际存在的边与所有可能存在的边的比例,网络的CC为网络中所有单个节点的CC平均值。通过比较模块网络和相同规模的随机网络,网络参数CPL和CC可以定义小世界网络[16],当其满足以下条件时:
CCsmall-world≫CCrandom
(5)
CPLsmall-world≥CPLrandom
(6)
其中,CCrandom和CPLrandom是1000个与目标网络相同规模的Erdos-Rényi(ER)随机网络的平均值。
进一步地,网络小世界指数被定义为[17]:
σSW=(CC/CCrandom)/(CPL/CPLrandom)
(7)
当网络的小世界指数σSW大于1时,该网络为小世界网络。
同时,基于小世界网络的生物学特性,对网络进行生物功能分析。通过DAVID(https://david.ncifcrf.gov/home.jsp)在线工具[18]对模块网络包含的基因进行GO(Gene ontology)功能富集的生物过程(Biological process, BP)分析。
1.5 生物标志物的选取及预后模型的构建方法
为了精确识别卵巢癌预后生物标志物,需要选择能够准确预测生存结局的数量最少的基因。根据网络节点的度(degree)、紧密中心度(Closeness centrality, CCL)、中介中心度(Betweenness centrality, BC)3个拓扑性质来衡量节点在网络中的重要性,对节点进行重要性排序。节点的度为该节点与其他节点直接连接的边数,CCL衡量了节点到其他所有节点之间的距离,BC用来衡量节点在网络信息传播中的作用。按以下公式计算节点的得分,对节点进行排序:
score=BC×CCL×log2degree
(8)
根据score选出得分最高的10个基因作为候选预后标志物,根据排名依次组合构建预测模型。预测模型的构建采用Cox比例风险模型进行,同时考虑各模型预测能力和模型基因个数,选择最优模型作为最终预测模型。利用Cox回归构建风险评分模型[19]如下:
(9)
其中N是候选预后标志物的数量,xi是预后标志物的表达水平,wi是Cox回归分析中候选预后标志物的回归系数。
根据风险评分Riskscore的中值将样本分为高低两类,运用KM(Kaplan-meier)图表估计生存函数,分析两组之间的预后差异。ROC(Receiver operating characteristic)曲线[20]和AUC(Area under curve)值被用来评价预后预测模型的效率,AUC值越大的分类器,预测模型的正确率越高。
2 结果及分析
2.1 卵巢癌预后相关基因
利用Cox回归对所有卵巢癌基因进行单因素回归分析,从17 779个基因中共筛选出了747个与OS显著相关的基因,称为卵巢癌预后相关基因。在这些预后相关基因中,一部分基因在先前研究中已被证实与卵巢癌预后相关,如:KLK4[21](HR=1.385;95%CI=1.04~1.85;logrankP=0.027)、CXCL9[22](HR=0.897;95%CI=0.83~0.97;logrankP=0.0034)。
2.2 构建的共表达网络及网络模块
为使得构建的共表达网络呈现无尺度网络分布,基于不同β条件下的log(k)与log(P(k))(k表示节点的连接度)的相关系数进行加权系数的选择。根据无尺度网络规则,加权系数β需满足log(k)与log(P(k))呈负相关,且相关系数越大,网络的无尺度特征越显著。最终选择相关系数大于0.9时,将共表达网络软阈值β设置为3(图1),此时网络节点度符合幂律分布,R2=0.89(图2)。在选定软阈值β=3后,将747个基因的相关性系数矩阵转换为邻接矩阵,进而转换为拓扑重叠矩阵,并运用层次聚类从中识别了一个预后模块(图3)。
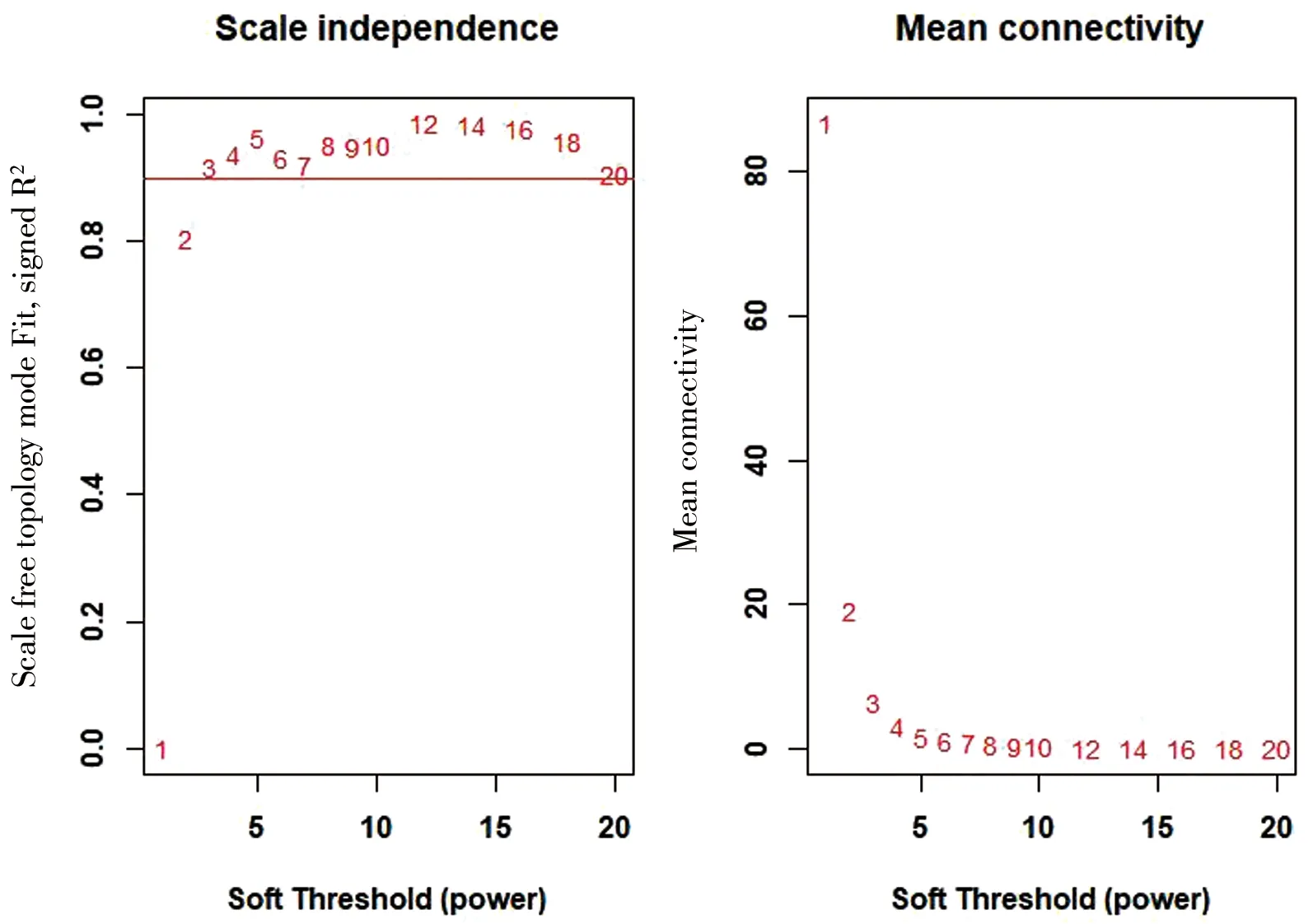
图1 确定加权系数β

图2 无尺度网络特性检验
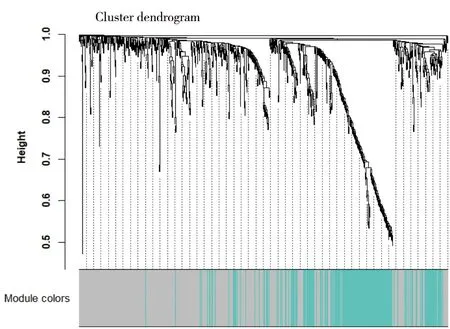
图3 基因聚类树状图
2.3 重加权网络拓扑分析及富集分析
对于基因共表达网络筛选得到的预后模块,用模块基因的蛋白质相互作用数据进行重新加权。从STRING数据库中下载得到了1154个模块基因的PPI,融合PPI信息后得到预后模块重加权共表达网络,该网络共包含96个节点和1768条边。进一步对网络进行拓扑分析,验证RMPCEN的小世界网络属性。拓扑分析表明,RMPCEN聚类系数为0.848,特征路径长度为1.614。同样节点数和边数的ER随机网络的平均聚类系数为0.389,平均特征路径长度为1.610。与ER随机网络相比,RMPCEN满足公式(5)和公式(6),因此RMPCEN符合小世界网络特性,其小世界指数为2.173。同时,输出RMPCEN中权重大于0.3的基因对,利用Cytoscape软件[23]绘制了RMPCEN的权重网络图(图4)。
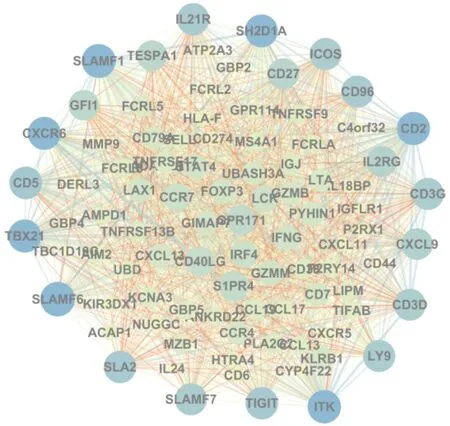
图4 预后模块重加权共表达网络
为了进一步验证RMPCEN在卵巢癌中的潜在功能意义,对RMPCEN中的基因进行了生物学过程富集分析。富集分析表明,RMPCEN中包含的262个基因在387条GO term中显著富集(FDR<0.05),FDR值小于1×10-10的生物学过程见表1。可以看出,RMPCEN中的基因集中参与了免疫系统过程、免疫反应、调节免疫系统过程等与免疫相关的生物学过程。
2.4 预后标志物的选取及预后模型的建立
计算RMPCEN中每个基因的score得分,并将基因按照score得分排序。考虑预后模型中基因的个数,选择了得分最高的10个基因测试其预测性能。排名前10的基因中,3个基因已被证实在卵巢癌的生长、侵袭、转移等过程中起重要作用,可用作卵巢癌治疗的良好靶点。这3个基因分别为:TBX21[24]、CXCR6[25-26]和TIGIT[27]。对于这10个候选预后标志物,根据score排名分别对于前n(1 表1 RMPCEN基因的生物学过程富集分析 表2 候选预后标志物及其构建的预后预测模型 同时考虑模型预测能力和模型基因个数,最终选择了前3个基因构建的预后模型,这3个基因分别为SLAMF6(HR=0.84;95%CI=0.71~0.99;logrankP=0.0416),SLAMF1(HR=0.63;95%CI=0.46~0.87;logrankP=0.005),CD2(HR=0.89; 95%CI=0.80~0.98; logrankP=0.0226)。预后模型由这3个预后标志物的表达值和Cox回归分析得出的回归系数线性组合构建:Riskscore=(-0.176 739 9×SLAMF6表达量)+(-0.459 172×SLAMF1表达量)+(-0.119 112 9×CD2表达量)。生存分析表明,高风险组(n=170)的患者生存率显著低于低风险组(n=169,logrankP=0.0055,见图5),这3个标志物能够较好地区分患者的预后情况。此外,模型的 ROC曲线显示,预后模型的AUC为0.689,表明了较好的模型预测性能。 图5 预后预测模型的KM生存曲线和ROC曲线 根据TCGA下载的数据构建了卵巢癌预后基因共表达网络,识别了网络的模块并利用模块基因的PPI数据对预后模块进行重新加权。得到的RMPCEN的小世界指数为2.173,符合小世界网络特性。这意味着,一方面,RMPCEN中的节点高度聚集:当一个节点连接到另外两个节点时,后两个节点也倾向于彼此直接连接;另一方面,网络中的平均最短路径长度几乎与随机网络一样低,从而说明RMPCEN具有高效的信息传递能力。因此,对RMPCEN中高度聚集的节点进行挖掘,通过score得分衡量节点的拓扑重要性。在score得分前10的基因中,TBX21[24]、CXCR6[25-26]和TIGIT[27]在卵巢癌细胞增殖和肿瘤生长中具有重要功能,表明拓扑学上重要的基因往往在疾病中发挥关键作用。目前,其余7个基因在卵巢癌中的功能尚未得到证实,但已被证明与癌症相关。ITK在大多数转移性黑色素瘤中异常表达,表明ITK抑制剂可能对黑素瘤治疗有效[28]。IL21R在乳腺癌细胞的增殖、迁移和侵袭中发挥作用[29]。因此,这些基因可能是卵巢癌的潜在疾病基因,值得进一步研究。 同时,对RMPCEN中的基因进行了GO富集分析,预测了与卵巢癌预后相关的潜在生物过程。生物学过程富集分析表明,RMPCEN中的基因集中参与了免疫系统过程、免疫反应、调节免疫系统过程等与免疫相关的生物学过程,这与先前的研究成果相符。目前,越来越多的证据表明,卵巢癌本质上是免疫原性肿瘤。流行病学和临床数据证明,卵巢癌患者的生存期与自发抗肿瘤免疫反应、肿瘤免疫逃逸机制相关。试验数据也证实了免疫疗法的功效,这将为卵巢癌免疫疗法提供新思路[30]。 本文在全基因组水平上对卵巢癌基因表达数据进行了系统的分析,识别了具有预后价值的生物标志物。以上讨论表明,通过整合PPI数据进行基因共表达网络分析有助于选择具有生物学意义的预后标志物。此外,拓扑上重要的基因在疾病的发展中表现出一致的表达变异,在疾病的发展中发挥关键作用。通过此方法挑选的预后生物标志物具有一定的可信度,可作为卵巢癌治疗的潜在靶点。

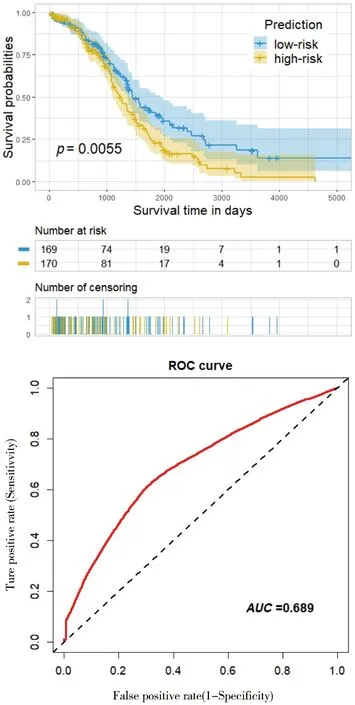
3 讨论与结论
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
安徽农业科学(2022年1期)2022-02-14
昆明医科大学学报(2020年12期)2021-01-26
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
中国癌症杂志(2015年4期)2015-12-09
肿瘤预防与治疗(2015年2期)2015-09-26
医学研究杂志(2015年7期)2015-06-22
医学研究杂志(2015年4期)2015-06-10
