
头孢菌素酰化酶高β-折叠区保守氨基酸附近位点突变研究
2020-10-20 03:04王继莲马刘峰
生物学杂志 2020年5期
任 羽, 常 瑞, 王继莲, 马刘峰, 王 东
(喀什大学 叶尔羌绿洲生态与生物资源研究重点实验室, 喀什 844000)
头孢菌素酰化酶(Cephalosporin acylase)能够一步催化头孢菌素C(Cephalosporin C,简称CPC)生成7-氨基头孢烷酸(7-Aminocephalosporanic acid,简称7-ACA),后者是医药工业生产头孢菌素的中间体。但头孢菌素酰化酶通过一步酶法对CPC的催化活性低,远未达到工业生产的要求[1]。因此,有必要对头孢菌素酰化酶(CPCase)进行分子改造,以使其适应工业生产需要。
头孢菌素酰化酶是通过间隔肽连接α-亚基和β-亚基而构成的二聚体蛋白,各国学者已针对β-亚基如何与CPC结合并催化其脱酰生成7-ACA这一科学问题开展了各种研究。梅婷等[2]对CPCase分子中影响活性的Y150、Q220和F347氨基酸位点进行饱和突变的研究结果发现,突变体Y150W/Q220G/F347Y的比活力提高至原来的8.3倍。Ishii等[3]将Met269突变为Tyr,发现酰化酶对CPC的一步转化率提高为原来的2.5倍。Ren等[4-5]对酶的Hisβ70等关键活性位点进行了验证,发现能够获得活性为原来的2倍的突变酶。Tian 等[6]通过计算酶设计的方法发现,单点突变L154βF,Y167βF,L180βF及其组合L154βF/L180βF和L154βF/Y167βF/L180βF能够改善酶的稳定性和活性。刘新花等[19]通过氨基酸的温度因子(B因子)指导头孢菌素C酰化酶的分子改造,发现突变体R218Q和K226V均可提高CPC 酰化酶的稳定性和催化效率。基于理性设计的定点突变常用来改造酶蛋白的性质和研究蛋白质的特定位点,具有快速、直接、准确率高的特点,在酶和蛋白质的分子改造上应用广泛[7-9]。
本研究以来源于Pseudomonassp. SE83的CPCase基因序列为模板[10],对其基因进行全局优化设计[11],人工合成目的基因,并克隆至表达载体pET28a,克隆命名为WT。选择WT高β-折叠区保守氨基酸附近的25个位点进行突变,测定氨基酸替换后CPCase对CPC的一步转化酶活,以期筛选到高活性的CPCase,提高该酶裂解CPC产生7-ACA的效率。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 克隆宿主菌E.coliDH5α、表达宿主菌E.coliBL21(DE3)、大肠杆菌表达载体pET-28a由本实验室保存。
1.1.2 酶和试剂 蛋白胨和酵母提取物均来自OXOID公司,EcoRⅠ、Hind Ⅲ、BamHⅠ和SalⅠ等限制性内切酶购自TaKaRa公司,考马斯亮蓝G-250和考马斯亮蓝R-250购自北京鼎国昌盛生物技术有限公司,天根快速定点突变试剂盒KM101、异丙基-β-D-硫代吡喃半乳糖苷(IPTG)和X-gal购于北京天根生物公司,CPC标准品和7-ACA标准品均由上海榕柏生物公司提供,其他试剂均为分析纯。
1.1.3 仪器 T60型紫外分光光度仪,宁波新芝SCIENTZ-ⅡD超声波破碎仪,湘仪GL-21M高速冷冻离心机,北京六一DYY-6C型电泳电源和JUNYI JY04S-3C凝胶成像。
1.2 方法
1.2.1 CPCase基因的优化、合成和表达载体的构建
以来自Pseudomonassp. SE83的CPCase蛋白质序列为模板,交给北京普尔普乐生物公司完成基因优化和合成的工作。人工合成目的基因(2350 bp)后,以EcoRⅠ和Hind Ⅲ为双酶切位点,将其构建到克隆载体并转入宿主菌DH5α中。以BamHⅠ和SalⅠ为双酶切位点设计引物,PCR扩增目的基因,连接至表达载体pET28a上,获得重组质粒,将重组质粒转化E.coliBL21(DE3)感受态细胞,获得重组大肠杆菌,本文用WT表示。
1.2.2 序列比对 将重组子WT的氨基酸序列与来自PseudomonasSY-77、CAD、PseudomonasN176等菌株的头孢菌素酰化酶氨基酸序列进行相似性比较,N176与WT的相似性在97%,CAD与WT的相似性是38%,SY-77与WT的同源性只有26%,但高β-折叠区保守氨基酸是相同的(图1),在保守氨基酸附近选择25个突变位点,采用定点突变的方法分别突变为Ala。
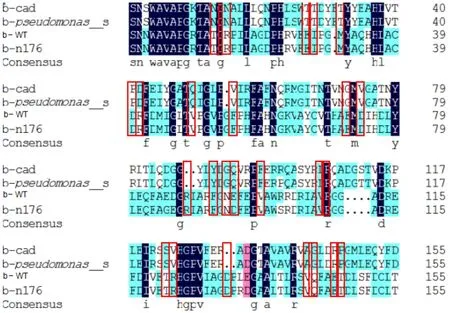
红框代表突变氨基酸
1.2.3 引物合成和突变基因表达载体构建 设计了25对突变引物,突变位点和引物序列见表1。以WT为模板,PCR反应体系、质粒模板的消化和转化宿主菌、突变克隆的筛选等步骤均按试剂盒KM101的操作方法进行,将突变克隆提取质粒,转化E.coliBL21(DE3)宿主菌,构建突变体,以突变位点命名,具体见表1。
1.2.4 CPCase的表达 将突变体接入含有50 μg/mL卡那霉素的LB液体培养基中,37 ℃,220 r/min振荡培养过夜,按1%接种量将过夜培养物转接入装有100 mL LB培养基的250 mL摇瓶,37 ℃,220 r/min振荡培养至OD600=0.6,加入终浓度0.5 mmol/L IPTG,22 ℃ 150 r/min培养12 h,SDS-PAGE检测目的蛋白表达情况。
1.2.5 CPCase活性检测 对蛋白粗提液进行酶活性测定,确定相对酶活。利用金属螯合亲和层析法(参考韦氏博慧公司的使用说明)对WT和F311酶蛋白进行纯化,考马斯亮蓝G-250定量,测定比活性和米氏参数kcat/Km。CPC转化的反应体系为20 mg/mL CPC钠盐500 μL(设置浓度梯度),酶液500 μL,37 ℃反应10 min,加入3 mL NaOH-冰醋酸终止反应,加入500 μLp-二甲基氨基苯甲醛(PDAB)室温显色15 min,于OD415下测定吸收值,同时利用高效液相色谱法进行测定。酶活单位定义为在37 ℃,pH 8.0时,每分钟生成1 μmoL的7-ACA所需的酶量。
1.2.6 分子建模与对接 利用Chemical Computing Group Inc.的Molecular Operating Environment (MOE)软件对WT和F311(F72βA)突变体进行分子建模,然后分别与CPC进行分子对接[12]。
2 结果与讨论
2.1 CPCase的表达和纯化

表1 突变引物设计(斜体部分为突变碱基)

M:分子质量标记(TaKaRa),其大小分别是200、116、97.2、66.4、44.3、 29.0和20.1 ku;B:牛血清白蛋白组分Ⅴ,分子量66.4 ku。图3同
将100 mL诱导表达培养物离心收集菌体,悬浮于10 mL Tris-HCl(pH 8.0)中,超声波破碎,离心取上清液,即为溶解的粗酶液,SDS-PAGE显示,WT为正常表达的未突变酶蛋白,两个亚基分别为58 ku和25 ku,I267和V288表达量很低,E266、F294、D313和V347没有完全剪切,形成83 ku的包涵体(图2),其他突变体都可正常表达并剪切为α、β 2个亚基,WT和F311的纯化结果见图3。
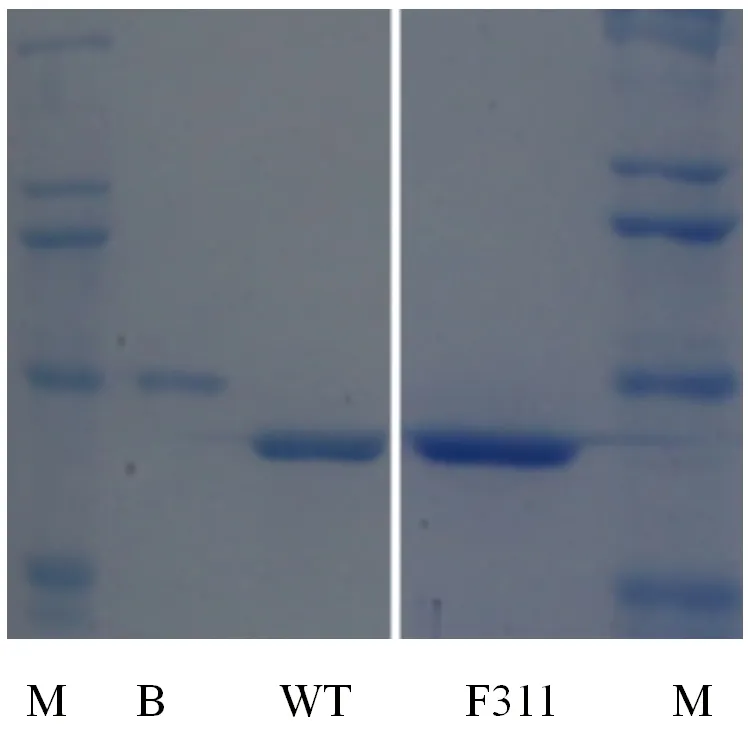
图3 CPCase重组表达纯化的SDS-PAGE分析
2.2 CPCase活性检测
以出发菌WT的活性为100%,测定突变体酶活性变化,结果发现,F311A的活性提高了10%,其他突变体的活性都不同程度降低,纯化后发现F311A的活性提高了50%,比酶活提高0.5倍,kcat/Km提高0.6倍(表2)。

表2 突变体的酶活
2.3 F311(F72β)位点突变分析
利用在线蛋白质结构预测软件SWISS-MODEL进行蛋白空间结构的同源模拟,结果(图4)发现本研究中的头孢菌素酰化酶的完整同源模型显示的活性位点与其他酰化酶的一致[13],使用Ser1β作为催化残基,这是N-末端水解酶的典型特征。Ser1β的羟基被保守的His23β固定,NH基团与His23β形成氢键。由于CPC羧基基团的作用,His70β骨架的NH 基团与 Asn242β的侧链形成氧洞。不同酰化酶[14-15]的催化残基是非常保守的,只有His70β(H309)可变,其作用是利用骨架来稳定氢键,与PGA 和CAD中相对应的残基作用是一致的。结合口袋是由CPC的羧酸盐(carboxylate) 基团的氧原子与Arg24β、Tyr32β 和 His57β互作形成的。CPC的氨基己二酸部分(Amino adipyl moiety) 通过与His178β形成氢键而被固定,后者同时与Asp177β发生作用。本研究中活性提高的突变位点是F311,即F72β,与活性位点处氨基酸His70β只隔1个丙氨酸。
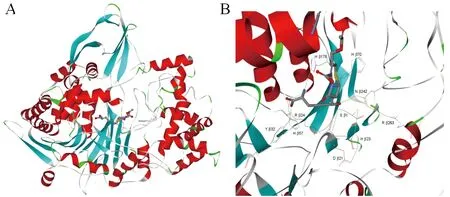
A: 酶的整体模型; B: 活性结合位点图

A: 表面观, Phe72是黄色突出; B: 3D互作模型
WT的Gln 373β能够与CPC形成氢键,这一结合被Phe72的侧链所阻碍(图5)。F72βA突变体的Arg 244β和Thr 256β 分别与CPC形成氢键,并且Phe 72β突变为Ala后,不会造成空间位阻(图6)。因此可以得出,Phe 72β 对CPCase发挥结合和催化活性有位阻效应,当Phe 72β突变为Ala 72β时,位阻效应消失,底物与酶的复合物在结合口袋内更加深入牢固,有利于酶的催化。
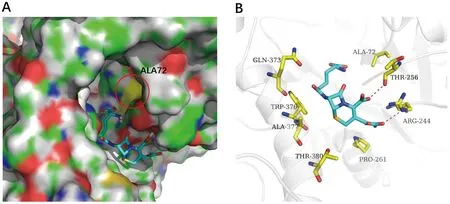
A: 表面观, Ala72是黄色突出; B: 3D互作模型
3 结论
对头孢菌素酰化酶高β-折叠区保守位点附近25个位点的可变氨基酸分别进行突变,得到活性提高50%的突变酶F311,比酶活和催化常数提高,分子对接结果发现,当Phe 72β突变为Ala 72β(F311A)时,位阻效应消失,底物与酶的复合物在结合口袋内更加深入牢固,有利于酶的催化。
本研究获得的突变酶F311可以再进行盒式突变,进一步阐述此关键位点的作用。此外,利用定向进化的方法,如易错PCR和DNA Shuffling技术也将获得更多活性提高的CPC酰化酶[16-18]。本研究为利用基因工程技术方法进一步提高CPCase活性提供了线索。
致谢:感谢中国农业科学院植物保护研究所张杰研究员课题组全体成员对本论文的载体构建和表达实验的无私帮助。
猜你喜欢
食管疾病(2022年1期)2022-11-26
中国药学药品知识仓库(2022年5期)2022-04-11
动物营养学报(2022年3期)2022-03-30
农业科技通讯(2021年1期)2021-03-06
天然产物研究与开发(2019年8期)2019-09-05
猪业科学(2018年5期)2018-07-17
上海农业学报(2017年3期)2017-04-10
天津医科大学学报(2015年2期)2015-12-22
医学研究杂志(2015年9期)2015-07-01
中国当代医药(2015年22期)2015-03-01
