
NR0B1基因新突变致先天性肾上腺发育不良1例
2020-10-20 12:42:00李江朱铭强黄轲董关萍
浙江医学 2020年19期
李江 朱铭强 黄轲 董关萍
患儿男,14岁2个月。因“发现生长缓慢14年余,阴茎短小4年余”于2019年7月16日至浙江大学医学院附属儿童医院就诊。患儿有一姐姐,现16岁,生长发育正常。患儿2011年在外院就诊,诊断为先天性肾上腺皮质增生症,未按时随访(具体资料不详),目前采用醋酸氢化可的松片20 mg/次、2次/d口服治疗,否认家族相关遗传代谢疾病史。入院查体:身高147 cm,体重34 kg,腰围68 cm,臀围74cm,体温36.3℃,脉搏92次/min,呼吸20 次 /min,血压 113/82 mmHg,神志清,身材匀称,无特殊外貌,齿龈、颊黏膜无色素斑片,心肺腹无殊,神经系统无阳性体征,四肢脊柱无殊,颜面部较黑,未见皮肤咖啡斑,双侧睾丸未触及,阴茎长4 cm,未见阴毛腋毛,无颈短颈蹼,无肘外翻。辅助检查:黄体生成素0.46 U/L,硫酸脱氢表雄酮<1.5 mg/L,睾酮3.09 nmol/L,雄烯二酮<0.30 nmol/L,血清双氢睾酮45.66 ng/L。甲胎蛋白 1.17 μg/L,癌胚抗原 0.61 μg/L。促甲状腺素 0.005 U/L,T32.17 nmol/L,T492.97 nmol/L,游离 T36.30 pmol/L,游离 T414.28 pmol/L,抗甲状腺过氧化物酶抗体1.06 U/ml,抗甲状腺球蛋白抗体1.76 U/ml。17α-羟孕酮0.1 nmol/L。促肾上腺皮质激素 410.0 ng/L,皮质醇<10 μg/L。立位血管紧张素Ⅰ4.54μg/L,立位血管紧张素Ⅱ225.20ng/L,立位肾素活性检测8.46μg/(L·h)。血气分析:钾4.4 mmol/L,钠 136 mmol/L,氯101 mmol/L。黄体生成素释放激素激发试验结果,见表1。人绒毛膜促性腺激素(hCG)激发试验:睾酮基础值<0.69 nmol/L,hCG激发后睾酮3.09 nmol/L。X线检查提示骨龄落后,骨密度低于同龄人。MRI检查示垂体高约3 mm。B超检查示双侧睾丸于腹股沟探及,双睾丸微石症;双侧肾上腺区未见明显异常。结合病史初步诊断:(1)性发育落后;(2)生长迟缓;(3)肾上腺皮质功能减退。在征得患儿家长同意后进一步完善基因检测,结果显示NR0B1基因第1外显子上存在c.864dupC(p.N289Qfs*10)半合子突变,见图1。该突变为移码突变,使得第289位上天冬酰胺(N)突变为谷氨酰胺(Q),在随后的10位变为终止氨基酸,导致多肽链提前终止,蛋白功能受损,多项蛋白功能预测软件预测该突变为有害。结合临床资料和基因检测结果,修正诊断为先天性肾上腺发育不良。进一步完善父母基因检测,因患儿母亲已故而未能采样,仅作了父亲基因测序,见图1。
讨论先天性肾上腺发育不良是一种因NR0B1基因突变或完全缺失引起的X染色体连锁隐性遗传病。NR0B1基因又称剂量敏感-性逆转-肾上腺发育不良基因1(DAX-1),位于 X 染色体短臂(Xp21.3),包含2个外显子与1个内含子,编码细胞核受体超家族的一种孤儿蛋白-DAX-1蛋白。DAX-1蛋白是调控肾上腺发育的重要转录因子,也存在于垂体分泌促性腺激素的细胞和下丘脑核团中,参与调控肾上腺、性腺的发育及激素的合成分泌。自1994年首次报道,截至目前国内外共报道了约250种NR0B1基因突变,突变位点主要在基因编码区,或影响蛋白折叠,或改变蛋白结构,导致蛋白被截断,从而影响功能[1-2]。

表1 黄体生成素释放激素激发试验结果(U/L)
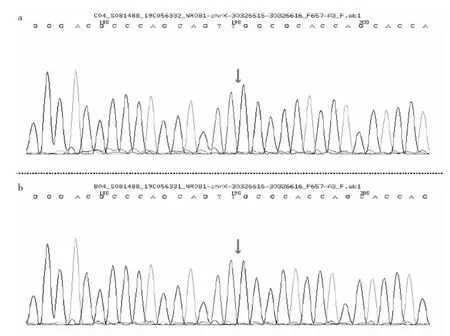
图1 先天性肾上腺发育不良患儿及其父亲基因测序结果(a:提示患儿NR0B1基因存在移码突变c.864dupC;b:提示其父相应位点无变异)
NR0B1基因突变可导致原发性糖盐皮质激素缺乏,先天性肾上腺发育不良患者多在婴幼儿时期表现为肾上腺皮质功能不全的失盐症状,随年龄增长,往往在青春期会伴随低促性腺激素性性腺功能减退(hypogonadotropic hypogonadism,HH)症状出现[3]。2007年上海瑞金医院首次在国内报道了此病,表兄弟2人首诊表现为典型的肾上腺皮质功能减退症,皮质激素替代治疗有效,后因青春期发育延迟查性激素水平及黄体生成素释放激素激发试验结果符合HH,进一步完善基因检测,发现2人基因均存在L262P突变,且各自母亲均为L262P突变,父亲均为正常野生型,符合X染色体连锁遗传方式,先天性肾上腺发育不良诊断明确[4]。检索中英文数据库,2007年至今关于NR0B1突变致先天性肾上腺发育不良的中国病例个例报道有13例,家系报道4篇,临床回顾性分析5篇。突变主要位于第1外显子,以移码突变及无义突变多见,与国外报道相似[5-6]。本研究中,NR0B1基因第1外显子上存在 c.864dupC(p.N289Qfs*10)半合子突变,该突变为移码突变,使得第289位上天冬酰胺(N)突变为谷氨酰胺(Q),其随后的10位变为终止氨基酸,多肽链提前终止,导致蛋白功能受损,理论上具有致病性。该移码突变为未见报道的新发突变,遗憾的是患儿母亲已去世,及家族中其他成员拒绝基因检测,未能完善家系检查,无法判断该突变来自母系遗传还是自发突变。
先天性肾上腺发育不良的临床表现复杂,早发病型多在生后几个月内发病,表现为体重不增或下降、呕吐、脱水、皮肤色素沉着等,常被误诊为先天性肾上腺皮质增生症、原发性醛固酮减少症、Addison病等。Flint等[7]报道1例21日龄新生儿,其临床表现酷似先天性肾上腺皮质增生症,且17羟孕酮轻度升高,11-去氧皮质酮明显升高。Iughetti等[8]报道了1例新生儿起病,仅表现为喂养困难,伴严重低钠、高钾、低氯,高肾素、低醛固酮,而皮质醇水平正常,促肾上腺皮质激素略有增加,考虑醛固酮减少症。两例患儿均依靠基因检测明确先天性肾上腺发育不良的诊断。2014年浙江省报道了1例8岁男孩因吐泻、腹痛、皮肤色素沉着,血检示高钾低钠、皮质醇降低、促肾上腺皮质激素升高、17-羟孕酮正常,诊断为Addison病,直至12岁仍性腺发育不良,经基因检测发现NR0B1基因E1半合子突变,最后诊断为先天性肾上腺发育不良[9]。本例患儿自6岁诊断为先天性肾上腺皮质增生症起,予以口服氢化可的松治疗,失盐等症状好转,本次就诊发现其性腺未发育、骨龄落后,17α羟孕酮多次检测均正常,考虑先天性肾上腺皮质增生症可能性不大,进一步完善基因检测后确诊为先天性肾上腺发育不良。因此,对年龄较大且出现肾上腺功能减退的患儿,应注意可能是先天性肾上腺发育不良,必要时行基因检测明确诊断。
NR0B1基因可能通过影响促性腺激素释放激素的合成分泌,从而影响青春启动与性发育,或通过类固醇生成因子-1影响下丘脑和垂体分泌促性腺细胞的发育,具体机制仍不明。少数先天性肾上腺发育不良患者在青春期晚期甚至成年后发病,称晚发型,占40%,临床表现往往不典型,HH可以是疾病的首发表现或唯一表现[10]。已有报道证实NR0B1基因型与表现型无明显相关性,即使同一家系、同一位点、同一突变类型,家族成员临床表现不尽相同[11-12],甚至有的表现为性发育不良,有的表现为性早熟[13]。目前为止,国外有16例次NR0B1基因突变致先天性肾上腺发育不良伴发性早熟的案例报道[14],视为下丘脑-垂体-性腺轴调控异常的另一类表现。国内首个NR0B1新突变合并中枢性性早熟的报道发表于2015年[15],患儿4月龄时以出现阴毛、阴茎及睾丸增大等性早熟症状发病,之前并无肾上腺皮质功能减退表现,经分子学检测证实为新的NR0B1无义突变(c.913C>T),诊断为先天性肾上腺发育不良。
综上所述,先天性肾上腺发育不良临床表现多样,对于有肾上腺皮质功能减退症状的患者,应评估其性发育情况,完善NR0B1基因检测,避免出现漏诊、误诊。此外,本例报道通过外显子测序发现的移码突变c.864dupC丰富了NR0B1基因突变谱,为先天性肾上腺发育不良的分子诊断提供了新的基因突变位点。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
成都医学院学报(2021年2期)2021-07-19 08:35:28
中国生殖健康(2020年2期)2021-01-18 02:51:26
小资CHIC!ELEGANCE(2021年46期)2021-01-11 05:24:50
睿士(2020年11期)2020-11-16 02:12:27
小学生导刊(2018年13期)2018-06-29 03:49:00
哈尔滨医药(2016年1期)2017-01-15 13:43:18
中国药业(2014年4期)2014-05-09 08:48:33
西部中医药(2014年6期)2014-03-11 16:07:43
河南医学研究(2014年5期)2014-02-27 14:52:41
