
DYT1型头颈部肌张力障碍及其颈椎病变并发症1例报告并文献复习
2020-10-15 08:20尚天玲沈延鑫郭春杰
中风与神经疾病杂志 2020年9期
尚天玲, 沈延鑫, 郭春杰, 孙 莉
肌张力障碍是一种持续性或间歇性肌肉收缩所导致的异常重复运动或姿势,或两者兼之的运动障碍[1]。2008版《肌张力障碍诊断与治疗指南》指出肌张力障碍按照病因分为原发性、肌张力障碍叠加、遗传变性疾病、发作性肌张力障碍和继发性,按照年龄可分为早发型(<26岁)和晚发型(>26岁)。TOR1A基因突变引起的DYT1型肌张力障碍属于原发性肌张力障碍,其早发型主要见于儿童及青少年,从四肢起病,逐渐累及全身其他部位,发展为全身型或多灶型;晚发型多为局灶型病变,可累及颈部及躯干,头面部受累罕见[2],累计头颈部更为少见。
1 临床资料
患者,男,62岁。因头及舌抖动30余年,右侧肢体活动不灵25 d入院。患者于入院前30余年无明显诱因出现头及舌抖动,呈持续性,与体位无关,夜间睡眠时减轻,病程中震颤未加重,曾多次就诊,未查明原因。于入院前25 d出现右侧肢体活动不灵,表现为右侧上肢尚可抬起,右侧下肢行走费力,就诊于当地医院行相关检查并给予治疗(具体不详)后患者症状无明显缓解,遂就诊于我院,门诊以“脑梗死”收入院。病程中伴有全身乏力、颈部僵硬疼痛,偶有排尿困难,无吞咽困难、饮水呛咳、构音障碍,无头晕、头痛,无视物旋转及不清。饮食睡眠尚可,二便正常,近期体重未见明显增减。既往史:高血压病史7 y,血压最高达180/100 mmHg,控制不详;否认抗精神病类药物及接触史;否认头颈部外伤史;吸烟40 y,20支/d,已戒2 m。入院查体:血压141/96 mmHg,脊柱侧弯,双肩不等高,头及舌可见震颤,右手掌骨间肌萎缩,双下肢肌容积较少,心肺腹查体未见异常。神清语明,咽反射减弱,左侧Horner(+),双侧Hoffmann征(+),右侧上肢肌力3级,右下肢肌力2级,左侧肌力正常,四肢肌张力正常,腱反射亢进,左侧T6水平以下痛觉减退,双侧Babinski征、Chaddock征阳性,余未见异常。
实验室检查:医学全外基因检测提示TOR1A基因常染色体显性遗传,杂合突变(见表1)。血常规、肝肾功、血尿酸、血同型半胱氨酸、空腹血糖、叶酸、维生素B12、铜蓝蛋白、甲状腺功能、传染病系列(肝炎、梅毒、艾滋)、肿瘤标志物等未见明显异常。

表1 医学全外基因检测结果
影像学检查:头部MRI平扫+弥散提示双侧多发腔隙性脑梗死、缺血灶(见图1);寰椎椎管狭窄,脊髓变细,脊髓水肿。磁共振脑血管成像(MRA):右侧椎动脉较对侧纤细,右侧大脑前动脉A1段未见显示,考虑先天发育可能。颈胸段MRI平扫(见图2):(1)颈胸段椎体骨质增生伴变性,所属椎间盘变性;(2)颈2~7、颈7~胸1椎体所属间盘后突;(3)颈4~5、5~6、6~7水平黄韧带肥厚或椎小关节骨质增生;(4)寰椎、枢椎层面后纵韧带肥厚,考虑继发寰椎层面椎管狭窄;(5)脊髓受压、水肿,所示层面脊髓中央管扩张。
综上,结合头、舌震颤病史及TOR1A基因常染色体显性遗传杂合突变,初步诊断为头颈部肌张力障碍。脊髓受压变形等影像学表现,考虑属于肌张力障碍引起颈椎病变并发症。结合高血压及吸烟等脑血管病危险因素、症状、体征及头颅核磁明确诊断为多发腔隙性脑梗死。综上,给予甘露醇脱水减轻脊髓水肿、替扎尼定缓解肌痉挛及营养神经、改善循环、抗血小板聚集等脑梗死常规治疗。寰椎层面脊髓受压明显,与脊柱外科商议后建议手术治疗,因考虑该部位手术风险高,家属选择保守治疗。出院时查体:右侧肢体肌力3级,余查体同前。出院后6 m、10 m分别电话随访患者,其未行脊柱手术治疗,仍有头及舌震颤,遗留有右侧肢体活动不灵(右上肢不能抬起,右下肢不能行走),出院后出现右侧肢体麻木,偶有大小便失禁。
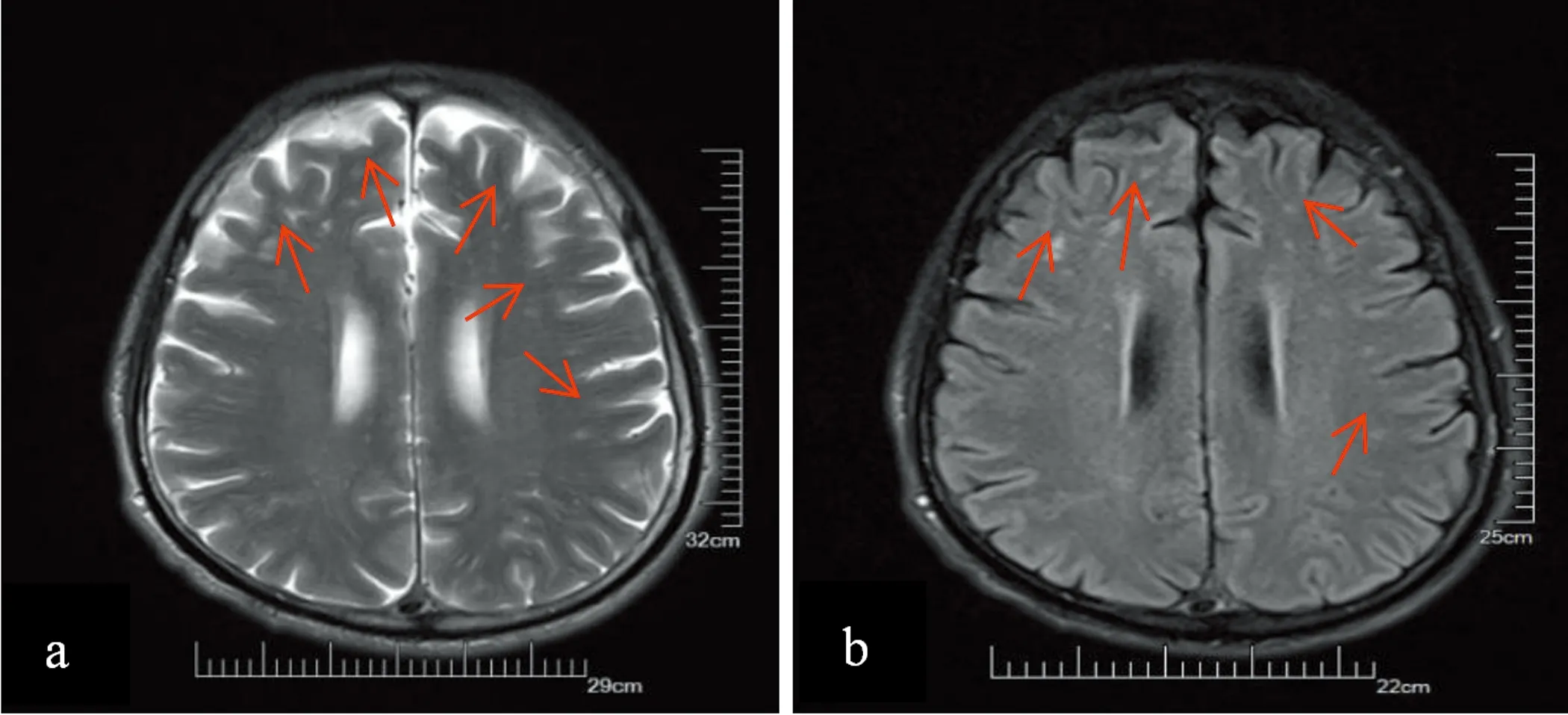
图1 头部MRI提示双侧多发性腔隙性脑梗死(a)T2WI呈高信号 (b)FLAIR呈高信号
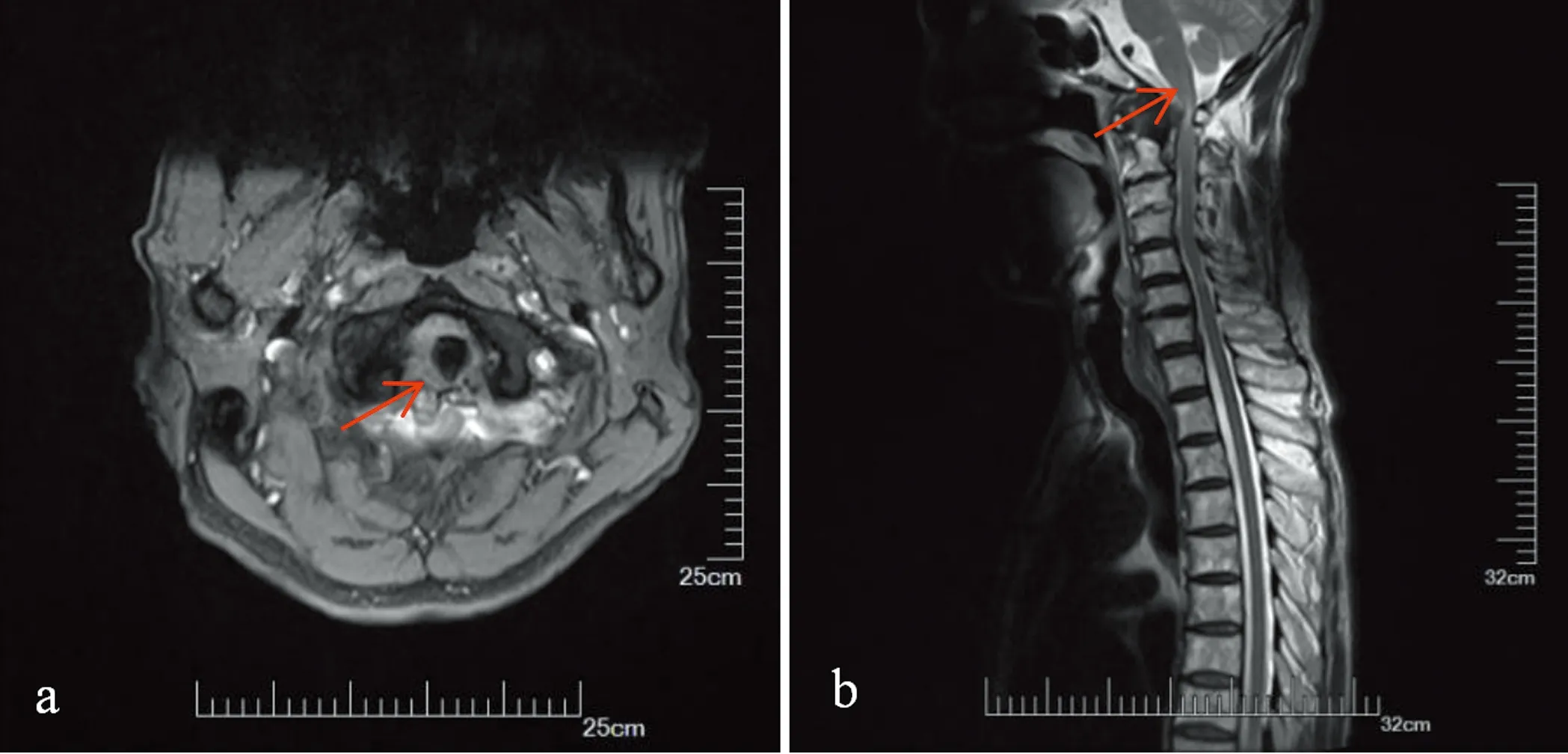
图2 颈胸段核磁提示颈椎病变(a)轴位T2WI:寰椎层面椎管狭窄,脊髓受压、水肿(b)矢状位T2WI:寰椎层面脊髓受压(红色箭头)
2 讨 论
DYT1型肌张力障碍是一种单基因遗传病,以常染色体显性遗传伴不同外显率为主,无明确的神经病理改变[3]。其诊断是通过分子遗传学测试鉴定杂合TOR1A致病性变异而建立,最常见的突变是第9号染色体5号外显子三联体密码子GAG的缺失(c. 904_906/907_909delGAG),几乎所有DYT1型肌张力障碍均可见[4,5]。DYT1型肌张力障碍以颈部起病者少见且累及颈部多为晚发型[6],我国大陆地区尚未报道累及头部肌肉者,出现头颈部肌张力障碍亦很罕见。
该患者头及舌震颤症状主要诊断为原发性头颈部肌张力障碍,原因如下:(1)排除继发性因素:首先排除帕金森病,症状方面:帕金森病震颤主要为手、手臂或腿等四肢静止性震颤,且随着年龄增加而加重,该患者震颤症状自年轻时发病持续至今,活动或休息时症状无明显波动,病程期间未加重且未出现帕金森病其他症状,如肌肉强直及运动迟缓等;基因方面:有报道[7]表明肌张力障碍合并帕金森症状基因检测有TAF1基因、CHC1/TH基因、ATP1A3基因、PRKRA基因,而该患者无上述基因突变。因此可排除帕金森病及肌张力障碍合并帕金森症状;其次,可排除肝豆核变性,眼科检查未发现角膜K-F环且肝肾功、铜蓝蛋白正常;再者可排除脑血管病,该患者合并多发腔隙性脑梗死,但脑梗死不能解释头和舌震颤症状,且震颤症状年轻时已出现,因此可通过详细询问病史,明确发病顺序相鉴别;最后,患者无头颈部外伤史及抗精神病类药物接触史,中年发病亦不考虑产伤等。(2)按照解剖结构将头和舌划分为头部,有报道[8]指出颈部肌张力障碍包括震颤型(单纯的头不自主震颤),因此该患者头部震颤可能为颈部肌张力障碍一种类型或单纯头部肌张力障碍,同时其伴有舌头震颤,舌肌肌张力障碍是一种局灶型肌张力障碍[9],因此头和舌震颤可划分为头颈部肌张力障碍。患者于30岁左右出现上述症状,考虑属于迟发型。行医学全外基因检测提示TOR1A基因有一个杂合突变:c. 907_909delGAG,导致氨基酸改变p. 303delE,为整码突变。根据ACMG指南,该变异提示致病性变异。因此,结合患者病史、症状体征及基因分析结果明确诊断为原发性迟发型头颈部肌张力障碍。根据基因型与表型之间的关系,TOR1A基因突变可引起头颈部肌张力障碍,但头颈部肌张力障碍是否只与TOR1A基因突变有关还有待研究。有报道[7]称DYT6/THAP1、DYT24/ANO3和DYT25/GNAL等均与颅颈段肌张力障碍相关,也有研究[10]表明DYT系列肌张力障碍排序证据尚不充足,且所有类型的肌张力障碍生物学机制都不一样,可能存在某种共同的分子机制。因此,尽管此病例基因检测提示TOR1A基因突变,但是否存在某种未知的导致头颈段肌张力障碍的分型尚不明确,今后可在分子学方面进一步探讨。
该患者存在右手掌骨间肌和双下肢肌萎缩以及肢体运动障碍(偏身运动障碍)、感觉障碍(偏身麻木、后颈部疼痛)、括约肌障碍(大小便失禁)等症状,其颈胸段核磁提示全颈椎椎体骨质增生及椎间盘凸出,周围韧带肥厚继发寰椎层面椎管狭窄。多发性腔隙性脑梗死不能完全解释上述神经缺损症状,考虑主要由寰椎椎管狭窄引起的继发性改变。寰椎属于颅颈交界区一部分,其常见病因有外伤、畸形、肿瘤、炎症等[11],类风湿关节炎、强直性脊柱炎等炎症性疾病可引起颈椎病变[12]。询问患者病史,其既往无头颈部外伤史,否认类风湿关节炎等病史,颈胸段核磁也未见肿瘤占位及扁平颅底、颅底凹陷、颅底压迹、寰枢椎失稳、脱位、半脱位、Chiari畸形等畸形,因此可排除上述病因。该患者查体可见颈椎侧弯、双肩不等高,结合影像学表现考虑颈椎病变由头颈部肌张力障碍引起。首先,颈椎病变可继发于肌张力障碍。有报道[13]称长期颈部肌张力障碍可导致上颈椎过早发生变性改变而导致颈椎不稳定,通过加重韧带变性过程促进颈椎病的发展。Doménech等[14]指出特发性颈肌张力障碍患者近1/3发展为脊柱侧弯,并且脊柱侧弯可以随着肌张力障碍得到治疗而控制。其次,有研究[15]表明孤立性脊柱侧弯可能是DYT1携带者肌张力障碍的表现特征。结合患者基因检测结果及临床表现,考虑该患者颈椎病变是TOR1A基因杂合突变导致DYT1型头颈部肌张力障碍的并发症,即头颈部肌肉长期不规则重复扭曲导致颈胸段周围韧带变性及颈椎侧弯并进一步导致椎间盘退变、脊髓受压等颈椎广泛性病变。
在现有检查条件下,TOR1A基因突变所致头颈部肌张力障碍及其颈椎病变的并发症都较为罕见,该个案可为肌张力障碍提供临床资料,但仍需要进一步考察和研究。此外,从该类疾病患者的临床诊治角度考虑,肌张力障碍患者在最初起病时,建议行相关基因检测,尽早诊断并积极地手术或药物治疗,以预防严重并发症的出现。
猜你喜欢
军事文摘(2022年8期)2022-11-03
现代仪器与医疗(2022年1期)2022-04-19
全科护理(2022年3期)2022-02-18
中国康复(2021年6期)2021-11-30
健康之家(2021年19期)2021-05-23
汽车工程(2021年12期)2021-03-08
中华养生保健(2020年5期)2020-11-16
电子制作(2019年7期)2019-04-25
家庭百事通·健康一点通(2017年8期)2017-08-18
中国民族民间医药·下半月(2014年5期)2014-12-02
