
热碱致魔芋胶与黄原胶共混凝胶的显微结构与流变规律
2020-10-15 08:51李培源李晓飞李安琪余文艳郭绰杨曦郭玉蓉
中国农业科学 2020年18期
李培源,李晓飞,李安琪,余文艳,郭绰,杨曦,郭玉蓉
(陕西师范大学食品工程与营养科学学院,西安 710119)
0 引言
【研究意义】魔芋为天南星科魔芋属植物,其球状块茎中主要成分为魔芋葡甘露聚糖,简称魔芋胶[1]。魔芋胶是一种性质优良的水溶性膳食纤维,不被人体的消化酶分解,但在小肠下部和大肠中被肠道菌群分解为葡萄糖和甘露糖,可调节人体膳食营养平衡[2-3]。魔芋胶凝胶食品长期以来一直被认为是健康和低热量的食品[4]。通常,魔芋胶在热碱处理下能形成热不可逆凝胶,但热碱所致的魔芋胶凝胶存在易析水、凝胶强度不足等问题,限制了魔芋胶凝胶在食品领域的进一步应用[5-8]。黄原胶是以玉米淀粉、蔗糖等为主要原料在甘蓝黑腐病野油菜黄单胞菌作用下,经好氧发酵产生的一种高黏度水溶性微生物胞外多糖[9-10]。黄原胶完全水化后可得到高黏度的溶液[11],但黄原胶在单独存在的情况下不能形成凝胶。有证据表明[12],魔芋胶与黄原胶在一定条件下复配能形成强度较高的凝胶,显现出良好的协同增效性。然而,这种凝胶是热可逆型凝胶,加热时,凝胶转变为溶胶,限制了这种凝胶在蒸煮、焙烤食品中的应用[13-16]。因此,探索魔芋胶和黄原胶复合体系在碱性条件下共热的凝胶化作用,以期将魔芋胶凝胶网络和魔芋胶与黄原胶协同凝胶网络合二为一,构建出兼具两种凝胶优点的复合凝胶,可为魔芋胶凝胶相关食品的设计提供理论依据和技术参考。【前人研究进展】ZHOU等[17]研究发现魔芋胶溶液与Na2CO3混合后并加热到70℃以上时,魔芋胶分子立即发生脱乙酰反应,诱导溶液发生凝胶化,进一步研究发现 KGM 链的分子间聚集不仅与氢键有关,还与疏水相互作用紧密相关。因此,降低魔芋胶凝胶温度时,疏水作用力减弱,魔芋胶凝胶强度也降低[18]。此外,由于乙酰基是赋予魔芋胶良好水溶性的关键因素,因此脱去乙酰基后,魔芋胶分子及其凝胶网络的持水能力大大减弱,引起凝胶析水[19]。迄今为止,已有少量研究致力于改善魔芋胶凝胶的这些缺点,包括增加魔芋胶浓度、优化凝胶形成条件等。然而,这些方法虽然可以起到一定的效果,但并未从根本上解决这一问题。近年来,有关混合多糖体系的研究已经得到迅速发展。与单一多糖体系相比,混合多糖体系的流变特性更具有可调控性[20]。一般地,可以通过调控混合多糖凝胶网络的形成和构建方式达到调整宏观凝胶体系物理特性的目的。例如,LI等[21]研究了葡萄糖酸内酯诱导的海藻酸钠和结冷胶混合体系的凝胶化作用,发现通过调整两种多糖各自凝胶网络的交联密度,可以实现整个凝胶体系物理特性的调控,包括凝胶硬度、溶胀性、热稳定性等。此外,李晓飞等[22]首次研究了碱性加热条件下,魔芋胶与黄原胶复合凝胶的形成过程,结果发现固定魔芋胶浓度为2.0%的前提下,在0—1.5%范围内随黄原胶添加量增加,复合凝胶破裂强度呈增加趋势,原因归结于热碱诱导的魔芋胶网络和黄原胶发生了协同结合作用。然而,有关热碱条件下魔芋胶和黄原胶协同结合的机理尚缺乏深入研究。【本研究切入点】本研究以魔芋胶和黄原胶复合体系为基质,研究热碱处理下魔芋胶凝胶网络的形成过程以及随后冷却过程中魔芋胶网络与黄原胶的协同结合作用,探讨热碱处理对魔芋胶和黄原胶复合体系最佳协同比例的影响。【拟解决的关键问题】揭示魔芋胶与黄原胶复合体系在热碱处理以及随后冷却过程中两种凝胶网络的形成过程及相互作用。同时,构建兼具两种凝胶优点的复合凝胶,为魔芋胶凝胶的物理改性提供理论依据和技术参考。
1 材料与方法
试验于 2019年在陕西师范大学食品科学与营养工程学院进行。
1.1 材料与试剂
魔芋胶和黄原胶均购自上海源叶生物科技有限公司,纯度为90%以上;无水碳酸钠、柠檬酸,购自天津市科密欧化学试剂有限公司,均为分析纯。
1.2 仪器与设备
电子天平 PL-203,梅特勒-托利多仪器(上海)有限公司;EYELA OSB-2100恒温水浴锅;电热鼓风干燥箱101-0A,天津心雨仪器仪表有限公司;TA. XT.Plus质构仪,英国stable micro system公司;环境扫描电子显微镜 Quanta 200,FEI公司;冷冻干燥机FD-1A-50,北京博医康实验仪器有限公司;AR-G2流变仪,美国TA公司。
1.3 方法
1.3.1 样品制备 准确称取25.0 g魔芋胶粉末溶解于1.0 L的去离子水中,一边添加魔芋胶粉末一边快速搅拌,随后将溶液置于90℃水浴锅中恒温加热4 h,并不断搅拌至魔芋胶完全溶解,得到 2.5%魔芋胶溶液。黄原胶母液制备方法同上。称取25.0 g黄原胶粉末溶解在1.0 L的去离子水中,一边添加黄原胶粉末一边快速搅拌,随后将溶液置于90℃水浴条件下不断搅拌至完全溶解[23]。
1.3.2 凝胶制备 在魔芋胶和黄原胶总浓度为2.0%的条件下,改变魔芋胶与黄原胶的比例,制备不同魔芋胶和黄原胶配比的复合体系。参照表1,两者总体积固定为160 mL,按照10∶0到0∶10的体积比改变魔芋胶母液和黄原胶母液的混合比例,之后各体系分别加入30 mL去离子水,并置于90℃水浴中加热搅拌至魔芋胶和黄原胶混合均匀。将4.0 g无水碳酸钠用10 mL去离子水溶解,缓慢加入到复合体系中,并快速搅拌,此时各个复合体系的总体积为200 mL。待复合体系混合均匀后,转入200 mL烧杯,封上PE保鲜膜,将烧杯在90℃水浴中加热2 h,取出烧杯,室温下自然冷却。室温平衡12 h后,测定各个凝胶的破裂强度,找到最佳配比[24]。采用该方法制备得到的复合凝胶命名为热碱协同处理的凝胶。
此外,本试验进一步制备了不同比例的魔芋胶和黄原胶复合体系,但该体系不在 90℃水浴条件下加热,即不诱导魔芋胶网络形成。参照表1,在总体积为160 mL的前提下,改变魔芋胶母液和黄原胶母液的体积比,制备不同混合比例的复合体系。将该复合体系转入200 mL烧杯中,置于90℃水浴中加热,并持续搅拌,使魔芋胶和黄原胶充分混合。各体系中分别加入30 mL温度为90℃的去离子水,继续搅拌均匀。之后,称取4.0 g碳酸钠粉末溶于10 mL去离子水中,充分溶解后,缓慢加入复合体系中,并剧烈搅拌,使体系混合均匀。立刻取出烧杯,自然冷却至室温。室温平衡12 h后,测定凝胶强度。这一方法制备得到的复合凝胶命名为碱处理凝胶。
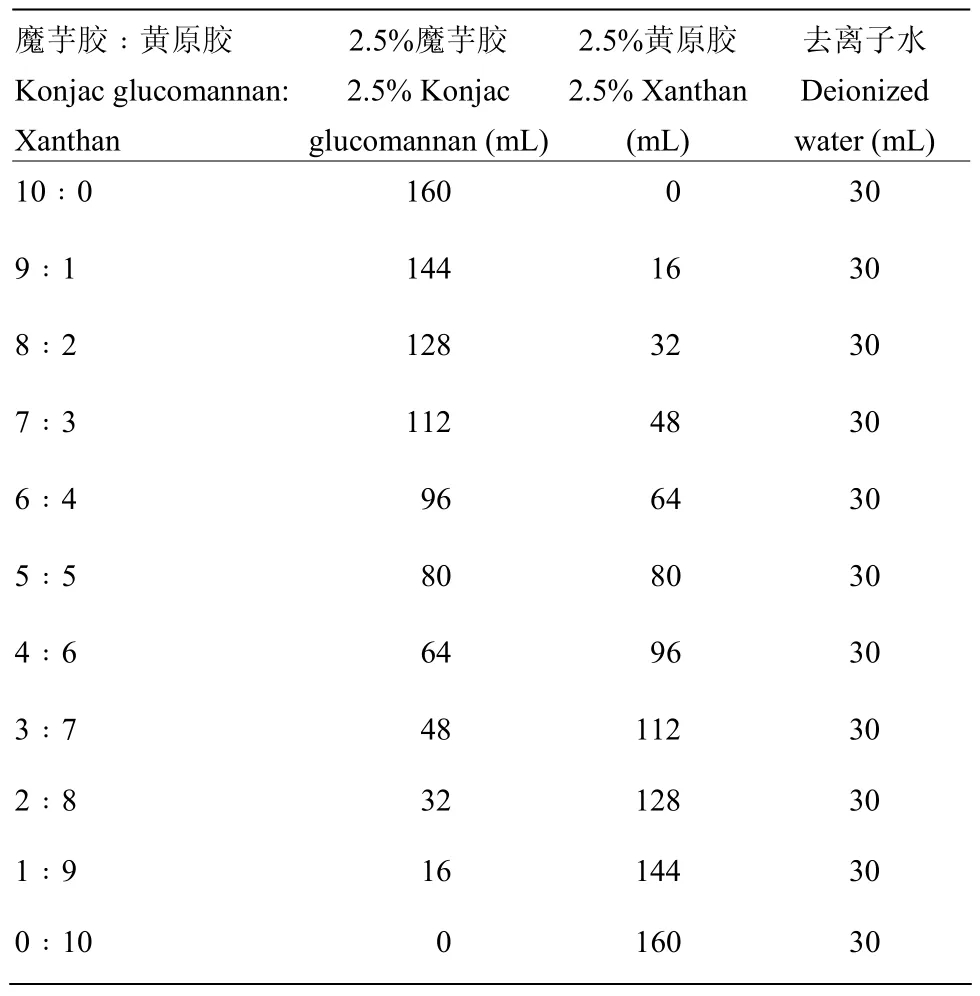
表1 样品制备各组分添加量Table 1 Addition amount of each component for sample preparation
1.3.3 凝胶破裂强度测定 凝胶平衡12 h后,采用质构仪测定凝胶破裂强度。参数设定为:TPA压缩模式,探头P 0.5(直径0.5 cm的圆柱状平头探头),测试前速度:1.0 mm·s-1;测试速度:1.0 mm·s-1;测试后速度:1.0 mm·s-1;感应力:2.0 g;凝胶破裂点所对应的力记为凝胶强度[25],每个测定重复3次。
1.3.4 凝胶微观形貌观察 参照上述步骤制备凝胶并于室温下平衡12 h后,将所有凝胶切割成边长约为2 cm的立方块,并迅速将凝胶浸没于液氮中速冻。液氮浸没约3 min后,将冻住的凝胶立刻转移至真空冷冻干燥机中,在-50℃条件下冻干48 h。将冻干的凝胶折断,断面喷金,采用Quanta 200 环境扫描电镜观察凝胶断面形貌,观察时选择高真空模式,高压为20.0 kV[26]。
1.3.5 溶胀特性 参照 1.3.2中的步骤制备复合凝胶。分别采用去离子水和2.0%的柠檬酸溶液浸泡(pH 2.06)复合凝胶。浸泡 12 h后取出凝胶,参照 1.3.3中的方法测定凝胶破裂强度。此外,参照1.3.4中的步骤,观测微观形貌[27]。
1.3.6 冻融稳定性 参照 1.3.2中的步骤制备复合凝胶,将所有复合凝胶置于-18℃冰箱中冷冻12 h,取出凝胶,室温下自然解冻6 h,参照1.3.3中的方法测定凝胶强度。此外,也测定冻融后凝胶的析水率。凝胶冷冻前的质量记为m1,凝胶冻融后将析出的水分除去,剩下的质量记录为m2。析水率可由下式计算:
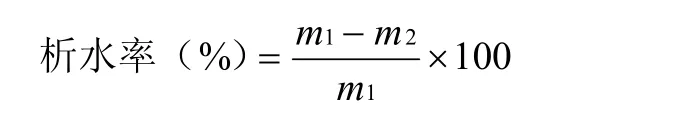
1.3.7 流变学性质 时间扫描:魔芋胶和黄原胶复合体系及碳酸钠溶液均按照1.3.2的方法制备并混合。按照表1添加配置好的2.5%魔芋胶母液、2.5%黄原胶母液和去离子水,并和 Na2CO3溶液混合,加入烧杯中快速搅拌均匀,制得魔芋胶与黄原胶比例分别为10∶0、9∶1、7∶3、5∶5的体系。随后,采用AR-G2型流变仪测定各复合体系的流变特性,所选夹具为直径为20 mm的糙面平行板。操作步骤如下:立刻移取2 mL混合溶液置于流变仪样品台上,平行板间距设置为 1 mm,并在平行板四周涂抹低密度硅油,防止测试过程中由于样品水分蒸发而造成较大误差。之后,在90℃条件下进行120 min的时间扫描,监测储能模量G'和损耗模量G''的变化趋势,应变范围为1%(线性粘弹区内),频率为1 Hz。
降温扫描:时间扫描程序结束后,立刻在90—20℃范围内进行降温扫描,降温速率为2℃·min-1,应变为1%,频率为1 Hz[28]。
频率扫描:当样品温度降低至20℃后,在该温度条件下平衡10 min,之后在1—100 rad·s-1频率范围内扫描,应变为1%。
方程拟合:为了进一步分析凝胶在相同碱浓度(2%)及温度(90℃)下的凝胶化过程,将上述时间扫描的G'汇总,并采用如下方程进行拟合[29]:

式中,G′sat为无限的加热时间下,G′的极限值;k为凝胶化速率参数;x为时间;y为储能模量G′。
1.3.8 数据处理 上述所有测定均重复3次,每次测试均需更换样品。所有图表均使用Origin 2018软件进行绘制,利用IBM SPSS22.0软件进行差异显著性分析,P<0.05表示差异显著。
2 结果
2.1 凝胶强度及外观
破裂强度是反映凝胶网络交联密集程度的重要指标。图 1-A为 2.0%的魔芋胶溶胶在热碱条件下(90℃)加热0—3 h的强度测定结果。如图所示,随加热时间增加,凝胶强度也逐渐增强,加热2 h后,凝胶强度虽然略有增加,但变化不显著(P>0.05)。由图1-B可知,加热时间为0 h时,魔芋胶不能形成凝胶,加热0.5 h后可形成明显的凝胶。基于这一结果,后续试验选择2 h作为凝胶热处理的时间。
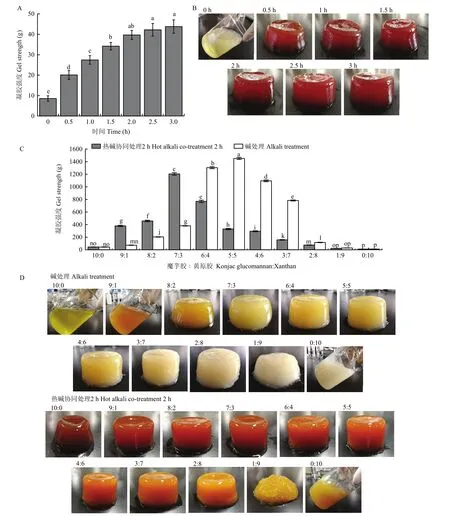
图1 不同魔芋胶和黄原胶比例复合凝胶强度及凝胶外观Fig. 1 Gel strength and gel appearance of different konjac glucomannan to xanthan ratios
将不同比例的魔芋胶和黄原胶混合,在热碱协同处理2 h和碱处理后(未在90℃条件下加热)分别测定凝胶强度,结果见图 1-C。如图所示,对于碱处理的凝胶,魔芋胶与黄原胶比例为5∶5时,凝胶破裂强度最高,约为1 400 g。对于热碱协同处理的凝胶,魔芋胶与黄原胶比例为7∶3时凝胶强度最大,约为1 200 g。可能是由于热碱协同处理过程中,多糖分子发生部分降解,影响了复合凝胶的强度[30]。魔芋胶与黄原胶比例为10∶0和9∶1时(图1-D、E),碱处理条件下凝胶无法形成,随黄原胶比例增加,凝胶呈现先增强后降低的趋势,和图1-C结果一致。热碱协同处理后,所有凝胶均呈棕色,且凝胶色泽随黄原胶比例增加逐渐变浅,当魔芋胶与黄原胶比例为1∶9和0∶10时无法形成凝胶,原因可能是黄原胶的存在降低了魔芋胶凝胶网络形成。
2.2 凝胶形貌
凝胶微观形貌与其强度密切相关[31]。由图2可知,不同比例的复合凝胶均形成了网络结构,但魔芋胶凝胶(魔芋胶∶黄原胶=10∶0)的微观形貌为层状结构,网络不明显;随黄原胶加入,凝胶开始出现网状结构,当魔芋胶与黄原胶比例为7∶3时,网络最为致密。当黄原胶比例继续增加时,网络结构变得疏松多孔。该结果与图1测得的结果相符,表明凝胶网络结构越致密,凝胶强度越大。
2.3 去离子水与柠檬酸溶液浸泡对凝胶结构的影响
由于凝胶制备过程中加入了2.0%的碳酸钠,可能造成凝胶存在明显的碱味,因此使用过程中需要将碱味去除。采用去离子水和柠檬酸溶液浸泡处理去除凝胶的碱味,并研究浸泡后凝胶强度及微观形貌的变化。如图3-A所示,去离子水浸泡后凝胶强度略有下降,但魔芋胶与黄原胶比例为7∶3时,凝胶仍具有最高的强度,表明去离子水浸泡并未改变复合凝胶的最佳协同比例。由图3-B可知,魔芋胶与黄原胶比例为7∶3时,凝胶网络结构最为致密。2.0%柠檬酸溶液浸泡后,魔芋胶与黄原胶比例为7∶3时凝胶强度也最大,且凝胶网络结构也最致密(图3-C、D)。然而,与去离子水浸泡的凝胶相比,柠檬酸浸泡后的凝胶网络更疏松,结构更松散,可能是因为柠檬酸与凝胶中的碳酸钠发生反应生成了二氧化碳,扰乱了凝胶原有的网络结构。
2.4 凝胶的冻融稳定性
将复合凝胶置于-18℃中冷冻12 h,取出后于室温下解冻,测定凝胶破裂强度、析水率及凝胶微观形貌。由图4-A、B可知,当魔芋胶与黄原胶比例为10∶0、9∶1和8∶2时,冻融后凝胶强度比冷冻之前有所上升,原因可能是冷冻过程中水分子结晶,迫使魔芋胶分子聚集形成稳定的交联,室温条件下解冻时,交联也不会解聚,引起凝胶强度增加。然而,魔芋胶与黄原胶比例为7∶3、6∶4、5∶5时,冻融后凝胶强度比冷冻前有所下降,原因可能是黄原胶比例增加,凝胶已经形成了较强的网络结构,冷冻过程中由于水分结晶,破坏了凝胶的原有网络,造成凝胶强度降低。此外,随黄原胶比例增加,凝胶析水作用逐渐降低(图4-C)。魔芋胶与黄原胶比例为10∶0和9∶1时,凝胶形貌为板状,网络结构不明显,魔芋胶与黄原胶比例为8∶2、7∶3、6∶4、5∶5时网络结构明显,表明魔芋胶比例越高,复合凝胶的冻融稳定性越差(图4-D)。
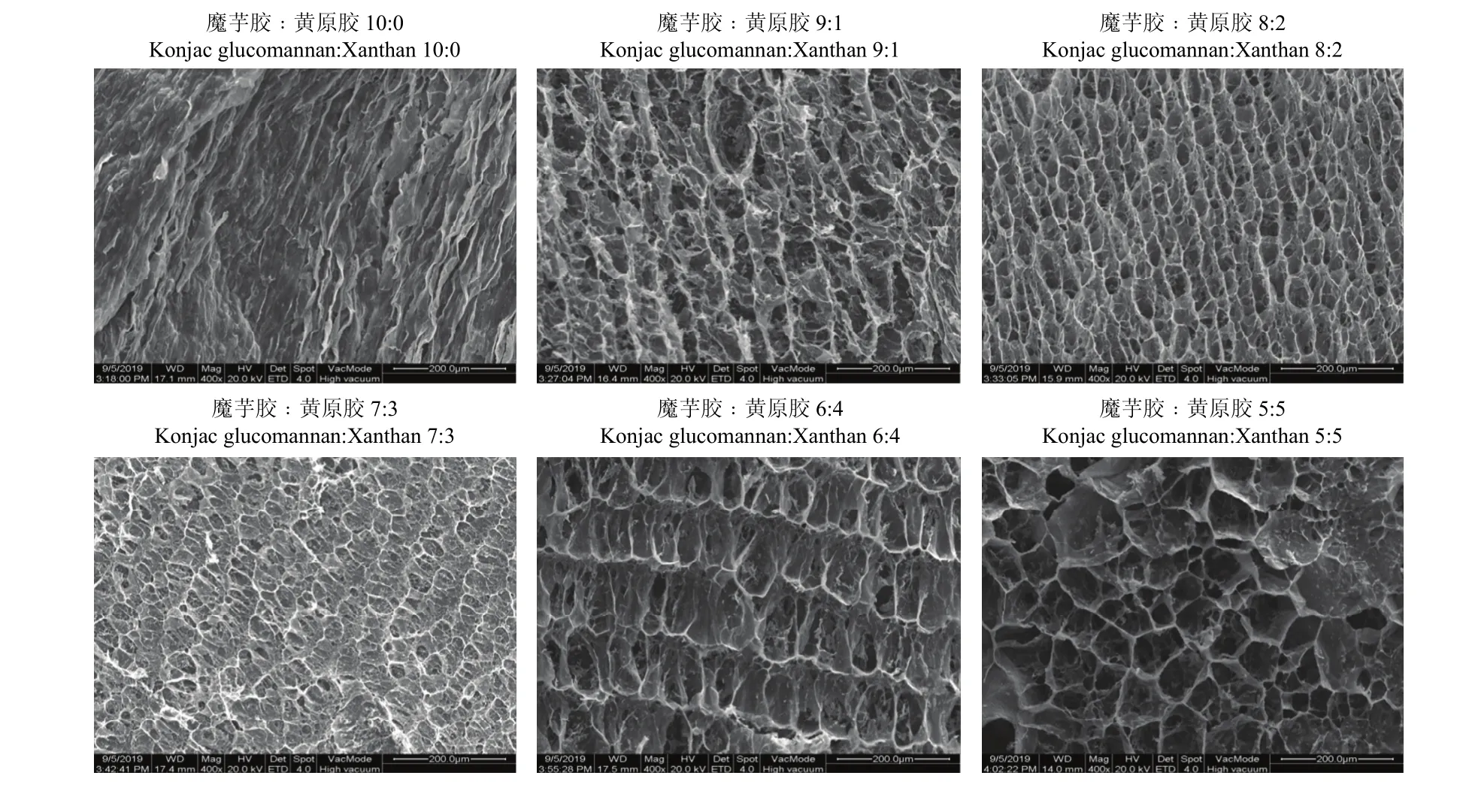
图2 不同魔芋胶和黄原胶比例对复合凝胶微观形貌的影响Fig. 2 SEM images of the composite gels with different konjac glucomannan to xanthan ratios
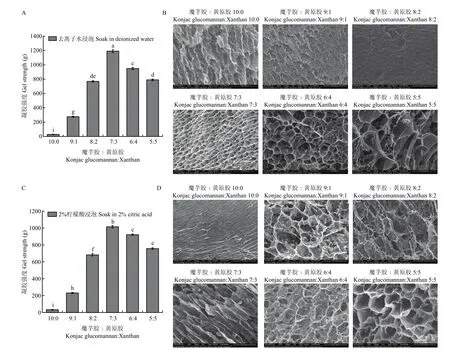
图3 水溶液浸泡对不同比例的魔芋胶和黄原胶复合凝胶破裂强度及微观形貌的影响Fig. 3 Effects of water immersion on the rupture strength and SEM images of the composite gels with different konjac glucomannan to xanthan ratios
2.5 流变特性
2.5.1 时间扫描 混合溶胶在90℃持续加热时,可观察到随时间增加,G'和 G''均呈显著增加趋势,0—1 200 s内G'增加速率显著高于G'',表明凝胶网络迅速生成。3 600 s之后,虽然G'和G''仍在继续增加,但增加趋势较为缓慢,说明凝胶结构发展比较完善。对于2.0%的魔芋胶体系,在240 s左右时G'和G''出现了交点,但在其他3个比例条件下,整个扫描过程中均未能观察到G'和G''的交点,可能是因为黄原胶比例增加后,由于黄原胶自身的分子间缠结作用,使得整个体系表现出 G'大于 G''的特征(图5)。
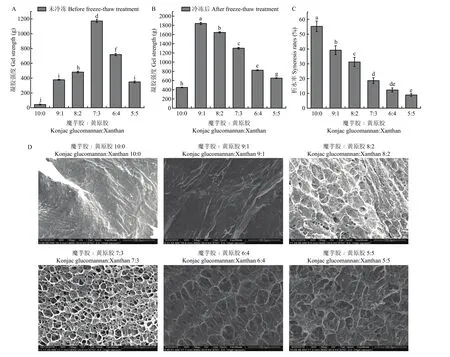
图4 冻融处理对不同比例的魔芋胶和黄原胶复合凝胶破裂强度、析水率及微观形貌的影响Fig. 4 Effects of freeze-thaw treatment on the rupture strength and syneresis rates and micro-morphologies of the composite gels with different konjac glucomannan to xanthan ratios
2.5.2 方程拟合 为了进一步分析凝胶在相同的碱浓度(2.0%)及温度(90℃)条件下的凝胶化过程,将上述时间扫描的 G' 汇总,并采用如下方程进行拟合[29]:,结果见图6。拟合的G'sat和k值见表2,所有拟合曲线的R2均在0.98以上,表现出较高的拟合精度。
由表2和图6可知,2.0%魔芋胶的G'sat和k值最大,随黄原胶添加量增加,G'sat和k都降低,说明黄原胶添加不仅降低了魔芋胶网络最终的强度,而且减缓了魔芋胶凝胶网络的形成速率。

表2 不同魔芋胶/黄原胶比例凝胶的G'一级动力学拟合方程参数Table 2 Parameters of the fitted equation of first order kinetics of G' of the composite gels with different konjac glucomannan to xanthan ratios
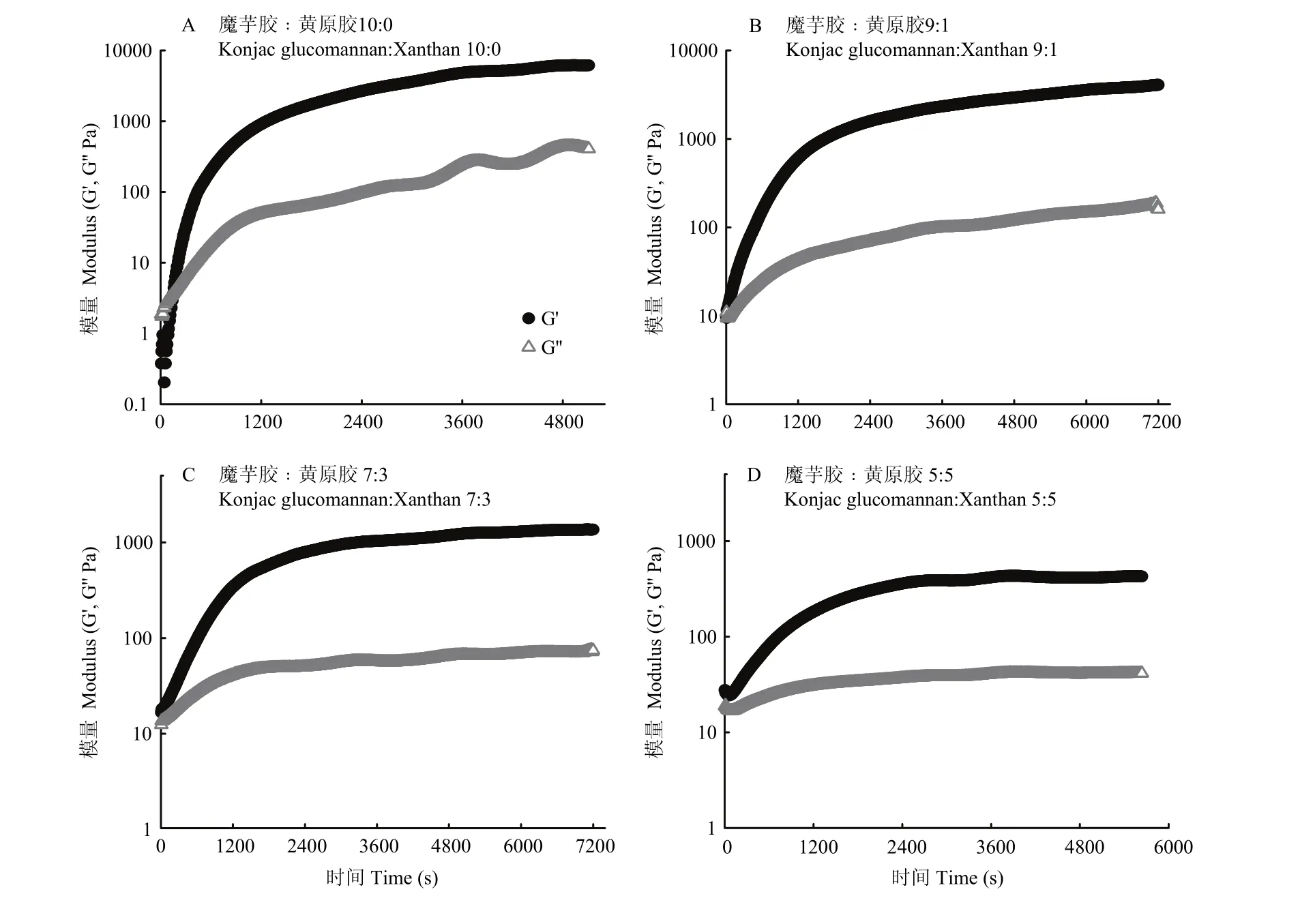
图5 不同魔芋胶和黄原胶比例的复合体系在90℃条件下时间扫描结果Fig. 5 Time sweep curves of the prepared konjac glucomannan/xanthan gels with fixed total gum concentration but varying konjac glucomannan to xanthan ratios at 90℃

图 6 不同魔芋胶和黄原胶混合比例的复合体系 G'的变化曲线及方程拟合Fig. 6 G' evolution process of the composite gels with different konjac glucomannan to xanthan ratios
2.5.3 温度扫描 魔芋胶与黄原胶协同凝胶作用与温度有关,热碱协同处理2 h后,2.0%的魔芋胶体系随温度降低,G'和G''呈不断下降趋势,说明魔芋胶凝胶强度也随之降低。由图7-B—D可知,G'在90—60℃范围内呈现降低趋势,60℃以后G'逐渐上升,可能是由于温度从90℃降低至60℃时,维系魔芋胶凝胶网络的疏水作用力变弱,从而导致凝胶网络弱化。60℃以后G'开始增加,可能是该温度下黄原胶和魔芋胶网络发生了协同结合作用,从而加固了凝胶体系,温度越低,协同结合作用越强[29]。当温度从 90℃降低至60℃时,G'表现出缓慢增加的趋势,然而当温度降低至60℃以后,G'迅速上升,表明黄原胶和魔芋胶协同作用发生的起始温度为60℃(图7-E)。这一结果也证实了热碱协同凝胶降温过程中G'的增加是由于黄原胶和魔芋胶网络发生了协同凝胶化。当温度从90℃降低至46℃时,G'呈现缓慢增加趋势,此后随温度降低,G'迅速上升(图7-F)。图7-E、F对比说明碳酸钠添加改变了魔芋胶与黄原胶凝胶体系协同作用的起始温度,在不加碱条件下协同化温度为46℃,而加入碱以后两者协同化温度上升至60℃。
2.5.4 频率扫描 频率扫描可得到凝胶储能模量 G'和损耗模量G''随振荡频率变化的关系,是评估凝胶状态的重要指标[32]。由图8可知,各比例条件下,凝胶体系在 1—10 rad·s-1的频率范围内模量几乎不随频率升高而增加,说明已经形成较强的凝胶。其中,魔芋胶与黄原胶比例为7∶3时,G'值最大,而魔芋胶与黄原胶比例为10∶0的凝胶G'最低,这一结果和凝胶强度测定结果一致。
3 讨论
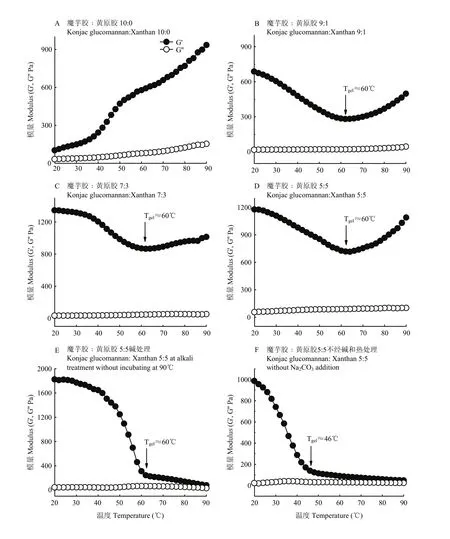
图7 不同魔芋胶和黄原胶比例下复合凝胶体系的温度扫描结果Fig. 7 Gel evolution process (G') of the composite gels with different konjac glucomannan/xanthan ratios during cooling
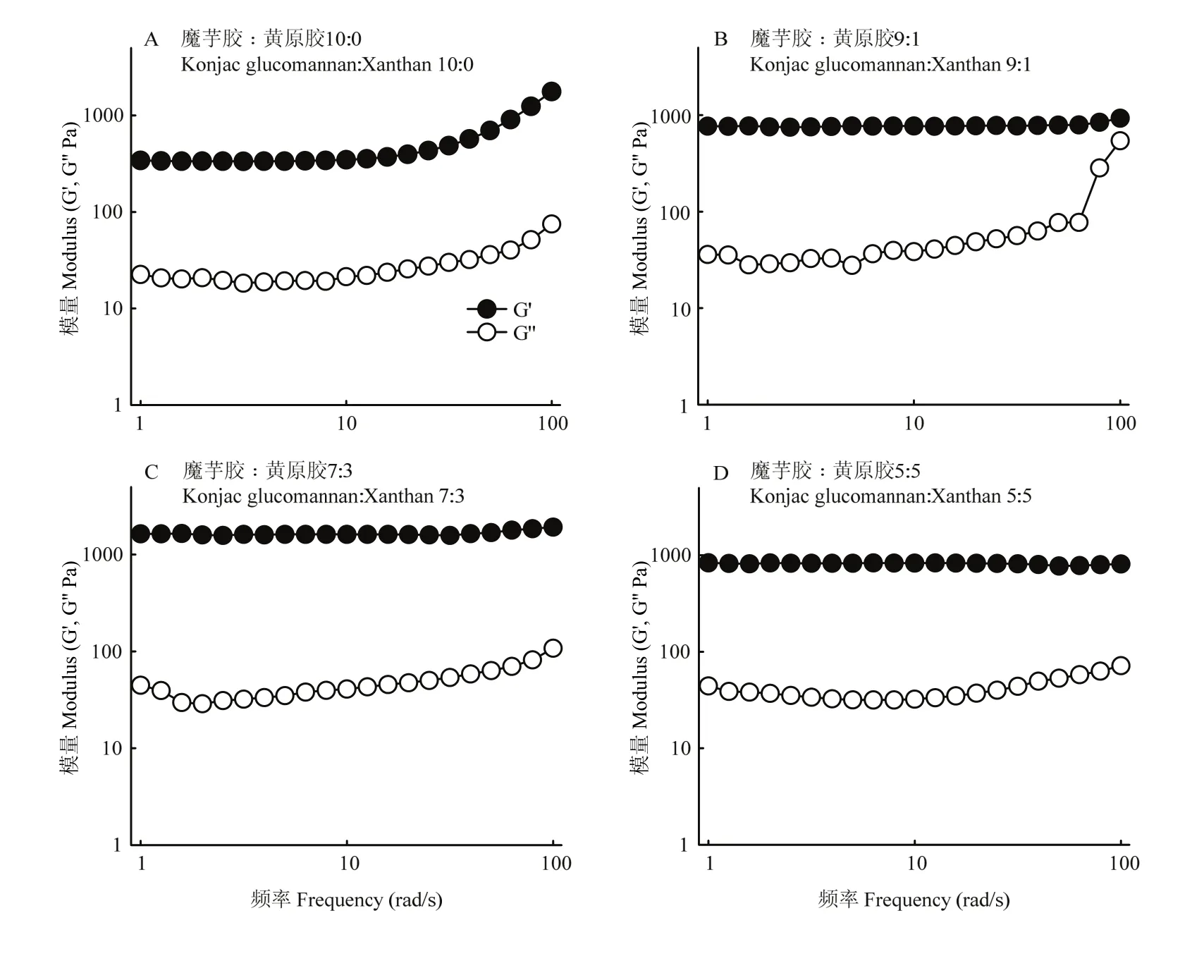
图8 不同魔芋胶和黄原胶比例下复合凝胶体系的频率扫描结果Fig. 8 Frequency sweep curves of the composite gels with different konjac glucomannan to xanthan ratios
研究表明[18],魔芋胶在碱性条件下形成凝胶的速率与魔芋胶浓度、碱浓度、加热温度、魔芋胶浓度与碱浓度之比有关。魔芋胶浓度越高,碱添加量越多、加热温度越高,魔芋胶去乙酰化速率越快,凝胶化速率也越高。本研究发现魔芋胶与黄原胶混合体系在最初的2 400 s内弹性模量和黏性模量均呈现迅速增加的趋势,2 400 s后,两者的增加速率逐渐变缓。然而,整个时间扫描过程中,基本未监测到G'和G''的交点,可能是由于混合体系加热温度较高,且碳酸钠添加量也较高,导致混合体系凝胶速率过快,在时间扫描程序开始运行之前,魔芋胶已经发生脱乙酰基反应,形成微弱的网络结构。因为G'是反应凝胶体系弹性特征的重要参数,因此本研究进一步通过方程拟合的方法探究热碱协同处理对魔芋胶与黄原胶混合体系的影响。结果发现,随魔芋胶与黄原胶比例降低,混合体系的凝胶速率常数k以及G'sat也降低,可能是体系中魔芋胶浓度降低所致。
加热2 h后,所有的混合体系均能形成凝胶。然而,当冷却混合凝胶体系至室温时,凝胶强度极大增加,显然是由于混合凝胶体系在冷却过程中,黄原胶分子与魔芋胶网络发生了协同作用[22]。因此,为进一步揭示这种协同作用的机制,本研究对混合凝胶体系进行降温扫描。结果发现,当温度从90℃降低至60℃时,所有的混合凝胶体系的G'均表现出降低的趋势,这可能是因为随温度降低,魔芋胶分子间的疏水作用力减弱,导致魔芋胶三维网络松散。然而,当温度进一步降低时,混合凝胶体系的G'表现出增加的趋势,表明黄原胶分子开始和魔芋胶网络协同结合,加固了凝胶网络。
黄原胶和魔芋胶最佳协同比例为4∶6(黄原胶∶魔芋胶),在此比例下,凝胶强度最大[24]。然而,黄原胶和魔芋胶最佳协同比例随体系中离子浓度的变化而轻微变化。例如,有报道显示[33],存在钠离子时,黄原胶与魔芋胶最佳协同比例由4∶6偏移至5∶5,这是由于钠离子屏蔽了黄原胶分子上微弱的负电荷,促使黄原胶分子发生聚集,减少了与魔芋胶分子发生协同结合作用的有效黄原胶含量,因此在达到最佳协同比例时需要更多的黄原胶。本研究发现,添加2.0%碳酸钠时,黄原胶与魔芋胶最佳协同比例为5∶5。然而,在90℃条件下加热魔芋胶和黄原胶混合体系2 h并冷却至室温后,最佳协同比例为7∶3(魔芋胶∶黄原胶)。原因可能是魔芋胶和黄原胶的协同结合位点数目有所变化。李晓飞等[22]研究发现,碱性条件下加热魔芋胶和黄原胶的混合体系并冷却时,魔芋胶分子首先发生脱乙酰基反应,形成魔芋胶凝胶网络,随后冷却过程中魔芋胶网络和黄原胶发生协同结合,进一步加固凝胶。因此,碱性条件加热时,魔芋胶网络首先形成,在随后的冷却过程中,与黄原胶协同结合位点明显减少,在达到最大协同比例时,需要更多的魔芋胶分子参与,意味着魔芋胶与黄原胶比例也相应增加。
4 结论
在90℃条件下加热魔芋胶和黄原胶共混体系时,形成热不可逆凝胶网络,当降低该体系的温度至60℃时,黄原胶分子与魔芋胶网络发生协同结合,增加了凝胶的弹性模量,随温度进一步降低,凝胶弹性模量持续增加。冷却至室温时,复合凝胶的最佳协同比例为魔芋胶∶黄原胶为 7∶3。经去离子水和 2.0%柠檬酸溶液浸泡后,凝胶强度均有所降低,但并未改变凝胶的最佳协同比例。冻融处理后,凝胶出现明显的析水现象,随黄原胶比例增加,凝胶析水作用降低。综上,本研究发现热、碱协同处理魔芋胶与黄原胶的混合体系可以改善单纯碱法诱导的魔芋胶凝胶冻融稳定性差、凝胶强度不足等缺点。
致谢:感谢陕西师范大学化学与化工学院大型仪器共享平台的仪器支持及管理老师在测样过程中的指导和帮助。
猜你喜欢
农业与技术(2022年10期)2022-11-19
中老年保健(2021年1期)2021-12-04
大连工业大学学报(2021年4期)2021-08-13
陶瓷学报(2021年1期)2021-04-13
发明与创新(2020年31期)2020-12-20
军事文摘(2020年20期)2020-11-16
小猕猴学习画刊·下半月(2020年8期)2020-07-28
家庭百事通·健康一点通(2018年3期)2018-03-30
实验流体力学(2018年5期)2018-02-13
农民致富之友(2017年23期)2018-01-02
