
IT15基因突变致亨廷顿舞蹈病一家系报告并文献复习
2020-09-24 08:48魏景景瞿千千吕海东
中风与神经疾病杂志 2020年8期
魏景景,韩 凯,瞿千千,吕海东
亨廷顿病(Huntington disease,HD)又称亨廷顿舞蹈病,是一种常染色体显性遗传性神经变性疾病,主要临床表现为慢性进行性舞蹈样不自主运动、精神障碍和痴呆。IT15基因为HD的致病性基因,IT15基因1号外显子内含有一段多态性三核苷酸(CAG)重复序列,当患者CAG重复拷贝数大于35次时即可引起发病[1]。我们收集1例IT15基因变异导致的亨廷顿舞蹈病家系,三代人中共有4例发病,本文对其临床特点、脑影像学改变和基因检测结果进行分析总结,并结合相关文献进行讨论。
1 资料和方法
1.1 临床特点 先证者,男,57岁,已婚,农民,初中毕业,汉族。因“精神行为异常19 y,伴不自主运动8 y”于2019年8月21日就诊。2001年患者出现头昏,工作效率低,伴入睡困难及多梦。当时头部CT未见异常,给予药物对症治疗。2003年,家属发现患者间断出现幻觉,经常说看到墙上爬着很多蚂蚁和一些不存在的人物,自己刻章去银行取钱等异常行为,遂至精神病院诊治,以“精神行为异常”口服舒必利和苯二氮卓类药物治疗。2005年以后逐渐出现打人骂人,多次就诊于我市精神病医院,以“精神分裂”加用奥氮平治疗,症状时轻时重。2010年患者逐渐出现双上肢舞蹈样不自主运动,行走不稳,重复性言语比较明显,夜间睡眠少,头部CT提示脑萎缩,口服奥氮平及劳拉西泮治疗,症状稍有改善。2017年以来患者舞蹈样不自主异常运动明显加重,不能独立行走,经常摔倒,同时伴有头部不自主扭动,不连贯的重复性言语,偶有饮水呛咳,睡眠困难,记忆力明显减退,2019年8月21日再次来我院就诊。既往发现糖尿病病史2 y,有吸烟饮酒史,现已戒烟酒10 y。查体:神志清,高级神经功能明显减退,言语构音不清,有重复性不连贯语言。头部可见不自主扭动,双侧瞳孔等大等圆,伸舌居中,四肢肌张力正常,腱反射活跃,肌力5级,四肢不自主舞蹈样抖动,独自站立和行走困难,双侧深浅感觉正常,病理征阴性。心肺腹查体无异常。家族史:父母非近亲结婚,父亲54岁时出现肢体舞蹈样不自主运动现象,69岁病故。母亲身体健康。兄妹3人,其中1妹妹已有类似症状,诊断为亨廷顿病。先证者的女儿有类似症状,但程度较轻(见图1)。
1.2 方法 (1)神经电生理:采用16导联数字化脑电图仪(上海诺诚Nation8128w),按国际10~20系统导联安放法则记录,设定敏感度100 uV/cm,低频滤波0.3 Hz,高频滤波20 Hz;(2)核磁共振:采用GE 3.0T核磁共振机对患者进行脑部平扫,扫描序列包括T1WI、T2WI、FLAIR及DWI序列;(3)心理检测:汉密顿焦虑量表(HAMA)、汉密顿抑郁量表(HAMD)、匹兹堡睡眠质量指数(PSQI);(4)基因检测:在先证者及其家属知情同意情况下,对先证者及近亲进行基因检测。采取静脉血4 ml,提取基因组DNA,进行基因遗传病筛查,由金域基因检验中心进行检测。
2 结 果
2.1 神经电生理 脑电图提示背景电活动为8~9 Hz的低幅α波,双侧波幅对称,调节、调幅不明显,光反应不明显。常规描记中及HV诱发3 min,各区可见低幅θ波,电压较低。结论为轻度异常脑电地形图。
2.2 脑核磁共振 幕上脑室显著扩大,中脑导水管形态结构大致正常,左侧壳核、右侧苍白球可见短T2低信号改变,双侧豆状核内见斑片状长T1、长T2信号影,双侧尾状核头部明显萎缩,双侧大脑脚萎缩,呈鸟嘴状改变,脚间窝池扩大,桥脑、延髓及双侧小脑形态结构未见明确异常(见图2)。
2.3 MMSE评分 14分。汉密顿焦虑量表(HAMA)评分:29分;汉密顿抑郁量表(HAMD)评分:24分;匹兹堡睡眠质量指数(PSQI):20分。
2.4 基因检测结果 先证者和其妹妹以及先证者的女儿均检测到IT15基因变异。先证者的IT15基因1号外显子的CAG重复数,一个为17次,属于正常范围;另一个CAG重复数为46次,属于全突变范围。其妹妹的IT15基因1号外显子的CAG重复数,一个为25次;另一个CAG重复数为46次。先证者女儿的IT15基因1号外显子的CAG重复数,一个为22次;另一个CAG重复数为55次(见图3)。
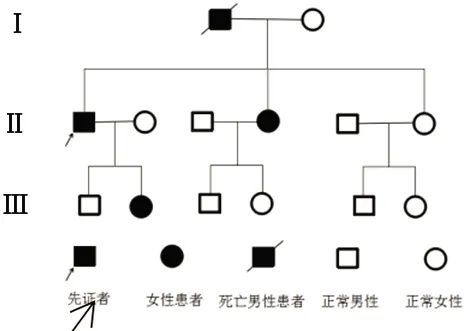
图1 先证者家系图谱
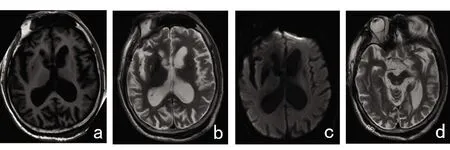
图2 a:T1Flair;b:T2;c:DWI:幕上脑室显著扩大,左侧壳核、右侧苍白球可见短T2低信号改变,双侧豆状核内见斑片状长T1、长T2信号影,双侧尾状核头部明显萎缩;d:T2双侧大脑脚萎缩,呈鸟嘴状改变,脚间窝池扩大
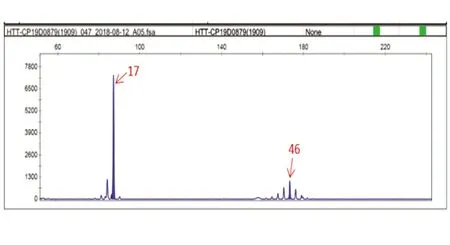
图3 先证者的IT15基因1号外显子的CAG重复数,一个为17次,属于正常范围;另一个CAG重复数为46次,属于全突变范围
3 讨 论
1842年由Waters最先报道描述了一种伴有精神异常的舞蹈样不自主运动性疾病。1872年美国医生George Huntington对该病进行了全面系统的描述,并指出其具有遗传性,遂以其名字命名为亨廷顿病。HD患者临床隐匿起病,以缓慢进展的舞蹈样不自主运动、精神异常和痴呆为临床3大特征。本病可见于各种族人群[2],欧洲人的患病率为5~7/10万,亚洲人的患病率较低,在日本约为0.5/10万,我国台湾地区的患病率约为0.1/10万。HD平均发病年龄为40岁,男女发病差异无统计学意义,发病后生存期一般为15~20 y[2]。本例先证者发病年龄为37岁,病情进行性加重,目前生活已经不能自理。先证者的妹妹45岁发病,目前尚可独立行走。
近年来,HD患者的非运动障碍症状越来越受到人们的关注。67%的HD患者存在睡眠障碍[3]。国外对于HD患者行为学障碍进行的临床研究显示[4,5],HD主要有抑郁(>55%)、淡漠(~30%)、易激惹(~10%)。本例先证者首先出现精神行为症状,先证者的妹妹也有淡漠、抑郁和睡眠异常等非运动障碍症状,说明精神异常和睡眠障碍是HD患者早期非常重要的非运动障碍症状。HD患者出现非运动障碍症状的机制尚不完全清楚,在正常情况下,亨廷顿病蛋白(htt)是在神经元的胞质中表达,但在HD患者脑组织的纹状体和大脑皮质等区域内的神经元核内可见异常的htt凝聚物,导致患者临床上出现神经功能障碍。
Farrer等研究[6]发现在HD患者中,约有10.5%的患者继发糖尿病,50%~60%患有糖耐量异常。HD患者继发糖尿病的确切机制尚不明确,有研究[7]认为突变的htt片段除了在神经元内形成聚集物外,也在内分泌细胞如胰岛和肾上腺髓质细胞中形成有聚集物,从而引起内分泌细胞损伤。本例先证者于2 y前发现糖尿病,口服二甲双胍0.25 g bid治疗,亦说明HD患者容易合并内分泌系统疾病。
HD的病理改变主要为双侧基底节和大脑皮质,所以头部CT及MRI检查可见大脑皮质和尾状核萎缩,以及脑室扩大[8],MRI较CT显示脑影像的改变更具优势。本例先证者MR提示尾状核萎缩和脑室扩大,呈全脑萎缩,符合HD的脑影像学改变。HD患者的脑电图呈弥漫性异常,临床上无明显特异性。
1993年Huntington病协作研究组[9]克隆了HD的致病基因IT15,并揭示了其第1号外显子内CAG重复序列的异常扩增是HD的遗传学基础。HD的临床表现和疾病进展与CAG重复序列和年龄有关[10],其拷贝数越多,发病年龄越早,临床症状越重。HD等位基因在传代过程中可出现子代CAG重复数目较上一代扩增的现象,使得子代发病年龄段提前。父系遗传的遗传早现更为明显,其原因可能与精子发生过程中存在不稳定的CAG重复序列扩增相关。本例先证者的父亲于54岁出现舞蹈样症状,先证者的发病年龄为37岁,先证者妹妹45岁发病,先证者的女儿30岁发病。本例家系的临床表型亦支持父系遗传的遗传早现,以及CAG拷贝数越多,患者发病年龄越早的观点。
临床上详细的神经系统检查和对认知功能、精神状态的评估,是诊断HD的基础。阳性家族史对其诊断具有重要意义,如果患者没有阳性家族史,或临床症状不典型,则基因检测是确诊的重要依据。1998年全美医学遗传学学会与全美人类遗传学协会Huntington病基因检测小组提出了基因诊断标准[1]:CAG重复数目≥40次时,为完全外显的Huntington病等位基因,携带者一定发病;CAG重复数目为36~39次时,则为不完全外显的Huntington病等位基因,仅部分携带者发病;CAG重复数目为27~35次时为正常等位基因,携带者不会发病。本例先证者和其妹妹IT15基因检测显示CAG重复数均为46次,先证者女儿的CAG重复数为53次,均为完全外显的Huntington病等位基因,所以先证者和其妹妹及其女儿均发病。
目前HD的临床治疗仍以经验性治疗为主[2,3,11],主要目标为控制症状、提高患者的生活质量。美国亨廷顿舞蹈病协会[11](HDSA)的治疗建议是强调HD的综合性治疗,药物治疗应与心理、社会和环境支持相协同,在疾病的不同阶段各有侧重。近年来,有学者从事HD的分子遗传学治疗研究,并且在动物模型实验中已显示出一定的疗效,期待着更多的研究为HD的治疗带来新的曙光。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
临床输血与检验(2022年3期)2022-06-22
中国生殖健康(2020年4期)2021-01-18
诊断学(理论与实践)(2020年1期)2020-04-28
肿瘤预防与治疗(2019年6期)2019-07-30
科学24小时(2019年4期)2019-06-10
郑州大学学报(医学版)(2019年3期)2019-06-03
医药前沿(2019年29期)2019-01-05
科学生活(2016年7期)2016-07-25
旅游世界(2014年11期)2014-12-05
