
酿酒酵母环核苷酸磷酸二酯酶1的异源表达、分离纯化与活性检测
2020-09-17 02:58李赤霞陈玉娟田元元韩潆仪王友升
食品科学 2020年18期
陈 滢,李赤霞,陈玉娟,田元元,张 萌,韩潆仪,王友升
(北京食品营养与人类健康高精尖创新中心,北京工商大学轻工科学技术学院,北京 100048)
环核苷酸磷酸二酯酶(phosphodiesterase,PDE)能够催化生物体内环核苷酸水解,通过调节生物体内环磷酸腺苷(cyclic adenosine monophosphate,cAMP)和腺嘌呤核糖核苷酸(adenosine monophosphate,AMP)的比浓度,进而调节由环核苷酸所调控的生物体内多种生理代谢过程[1]。该酶对细胞生命活动起着极其重要的作用。目前研究表明,人体内的PDE能够对心脑血管疾病[2-3]、炎症[4]、视觉传导[5]等生理性疾病起到调控和治疗的作用,于是PDE作为一种治疗靶点进入研究者的视线[6]。
酿酒酵母(Saccharomyces cerevisiae)在食品工业中有非常重要的地位,广泛应用于酿酒工业、发酵食品、能源化工业、酵母食品以及生物防治等领域[7-8]。如吴轩德等[9]利用醋酸杆菌优化S.cerevisiae在白酒的酿造中产生乙酸乙酯的过程;陈安特等[10]利用S.cerevisiae优化萝卜泡菜的发酵过程,证明S.cerevisiae能够抑制泡菜过度酸化,并能产生酯类香气;李强等[11]在桑葚中筛选出1 株S.cerevisiae,对葡萄采后的黑曲霉病有较好的防治效果。此外,基因组测序结果显示,S.cerevisiae与人类基因具有约23%的同源性,许多与人类细胞代谢相关的蛋白质如细胞周期蛋白、信号蛋白等都已在酵母中发现其相应的同源物[12-13]。
目前,已在S.cerevisiae中发现了两种PDE,分别是低亲和力的cAMP PDE1和高亲和力的cAMP PDE2[14]。其中有研究表明S.cerevisiae PDE1基因的缺失,对S.cerevisiae发酵的磨盘柿子果酒的抗氧化能力和挥发性物质有一定的影响[15]。但是,目前对于S.cerevisiaePDE1生物活性的报道较少,且对于如何获得大量高纯度的S.cerevisiaePDE1蛋白也没有相关报道。随着基因工程技术的发展,分离纯化技术的不成熟成为限制重组蛋白应用的主要因素[16]。
本研究选择大肠杆菌(Escherichia coli)作为表达载体,将聚合酶链式反应(polymerase chain reaction,PCR)扩增出的S.cerevisiae PDE1基因片段转入其中,诱导表达以获得大量的目的蛋白,并运用多种纯化方法对目的蛋白进行纯化。目前实验室中使用较为广泛的是亲和系统纯化,如邓学天等[17]应用Ni柱纯化其表达的TNF-α蛋白,获得较高纯度的目的蛋白;王博达等[18]通过Ni柱亲和层析获得4.6 倍纯度的解淀粉芽孢杆菌凝乳酶,且能够达到66.51%的回收率。Q-Sepharose纯化系统是应用离子交换原理进行蛋白纯化的一种手段,有学者应用Q-Sepharose纯化从白腐真菌发酵液中纯化一种新的漆酶,纯化效果良好[19]。分子筛纯化系统是利用分子质量的大小进行纯化的一种方式,金昊[20]应用Ni柱、离子交换柱和分子筛对MCUR1蛋白进行纯化,得到高纯度蛋白,且蛋白能够应用于蛋白结晶。本研究采用以上3 种不同的纯化系统,依次对表达的目的蛋白进行亲和层析、离子交换柱和分子筛的纯化,目的是得到高纯度的重组目的蛋白,并进一步研究S.cerevisiaePDE1蛋白的生物活性。
1 材料与方法
1.1 材料与试剂
1.1.1 菌体与质粒
野生型S.cerevisiae、E.coli感受态细胞BL21、DH5α、质粒pET28a和pGEM-T,均为本实验室保存。
1.1.2 试剂
T4 DNA连接酶、DNA聚合酶、NheI内切酶、EcoRI内切酶 美国Biolab公司;DNA凝胶电泳回收试剂盒(纯化回收DNA片段)、高纯度质粒小量提取试剂盒 美国Thermo公司;Ni-NTA agarose 德国Qiagen公司;Q-Sepharose Fast Flow、Sephacryl S200美国GE Healthcare公司;胰蛋白胨、酵母浸粉 中国北京奥博星生物技术有限公司;氯化钠 北京化工厂。
1.2 仪器与设备
French Press高压破碎仪 美国Glen Mills公司;层析柜 生工生物工程(上海)股份有限公司;JP61M/HV-85高压灭菌锅 日本Hirayama公司;恒温摇床 哈尔滨东联电子仪器厂;model868 pH计、A2净化工作台 美国Thermo公司;5810R高速离心机 德国Eppendorf公司。
1.3 方法
1.3.1 重组质粒载体的构建
根据S.cerevisiaePDE1基因序列(NCBI Gene ID:852644)设计引物,该基因编码序列全长为1 110 bp。上游引物序列:5’-TCGGCTAGCATGGTTGTATTCGAAAT AA-3’;下游引物序列:5’-TCGGAATTCTTATAGAAAC AAAGTGTGGC-3’。下划线序列分别为NheI和EcoRI的酶切位点,引物由生工生物工程(上海)股份有限公司合成。利用NheI和EcoRI限制性内切酶对pGM-T-S.cerevisiae PDE1进行双酶切并回收目的基因产物。T4连接酶将目的基因产物与pET28a质粒构建pET28a-S.cerevisiae PDE1重组质粒并进行DNA电泳纯化后双酶切验证。将重组质粒转入E.coliDH5α感受态细胞,并选取阳性单克隆菌在含有50 μg/mL卡那霉素的LB液体培养基中过夜培养,将菌液进行测序鉴定(UNC-CH Genome Analysis Facility)。
1.3.2 重组pET28a-S.cerevisiaePDE1质粒的诱导表达
将获得的pET28a-S.cerevisiae PDE1重组质粒热激法转入到E.coliBL21感受态细胞中,并接种到含有50 μg/mL卡那霉素和30 μg/mL氯霉素的LB固体培养基中,37 ℃培养12~16 h后,接种至含有50 μg/mL卡那霉素、30 μg/mL氯霉素和0.4%葡萄糖的2×YT培养基中,在37 ℃培养至A600nm为0.7,加入0.1 mmol/L异丙基硫代半乳糖苷低温诱导(15 ℃)48 h[21]。离心收集菌体。分别对诱导前、诱导24 h和48 h表达菌体测定A600nm值,并按照等菌体量进行取样,进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)。
1.3.3 蛋白纯化
1.3.3.1 菌体破碎
收集的表达菌体使用提取液按照1∶100的比例重新悬浮,French Press高压(压力1 200 psi)破碎菌体并对获得的细胞破碎液分别取样,即破碎总蛋白、离心上清蛋白和离心沉淀蛋白,并进行SDS-PAGE检测。
1.3.3.2 基于重组蛋白多重性质的分离纯化
将离心后的上清蛋白上样至Ni-NAT柱中进行吸附和洗脱并收集目的蛋白[22],将Ni-NAT柱纯化后的S.cerevisiaePDE1蛋白加入2.5 mmol/L CaCl2、0.5 U/mL牛凝血酶,25 ℃、150 r/min条件下酶切2 h,切除目的蛋白的His-Tag。将酶切后的蛋白样品进行Q-Sepharose分离纯化[23],并将收集的蛋白浓缩到10 mL以下再进行Sephacryl S200柱分离纯化[24],Ni-NAT、Q-Sepharose和Sephacryl S200分离纯化的蛋白均进行SDS-PAGE分析并测定其A280nm值,定义A280nm等于1时的蛋白质量浓度为1 mg/mL,最终蛋白浓缩至10 mg/mL并冷冻保存。
1.3.4S.cerevisiaePDE1蛋白酶活性测定
参照Wang Huanchen等[25]方法:98 μL assay buffer(20 mmol/L Tris-HCl,pH 7.5;4 mmol/L MnCl2,3H-cAMP(1∶100稀释),1 mol/L二硫苏糖醇)加2 μLS.cerevisiaePDE1蛋白,反应15 min后,加入0.2 mol/L ZnSO4溶液终止反应,再加入0.25 mol/L的Ba(OH)2溶液,离心10 min后,吸取上清液加入到30% ScintiSafe测定酶活性[25]。同时,测定5 个不同浓度下的S.cerevisiaePDE1蛋白对于cAMP和环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)的底物转化酶活性数据,按下式计算对两种底物的转化率:

1.4 数据统计与分析
采用凝胶成像仪对电泳图进行分析。采用GraphPad Prism 5进行非线性回归得到Michaelis-Menten方程曲线,并计算R2。
2 结果与分析
2.1 重组质粒载体的构建
S.cerevisiae PDE1PCR扩增的目的基因片段通过胶回收和酶切后,电泳结果见图1。可以看出目的基因PCR产物经酶切后在3 000 bp和1 000 bp左右均出现单一条带,其中3 000 bp处为载体pGM-T条带,1 000 bp处为目的基因条带,从结果中能够证明该PCR扩增产物分子质量符合预期,并且酶切成功。
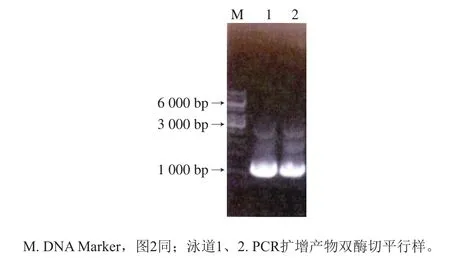
图1 S.cerevisiae PDE1基因PCR产物酶切后DNA电泳结果Fig.1 Electrophoresis of PCR products of S.cerevisiae PDE1 gene after enzymatic digestion
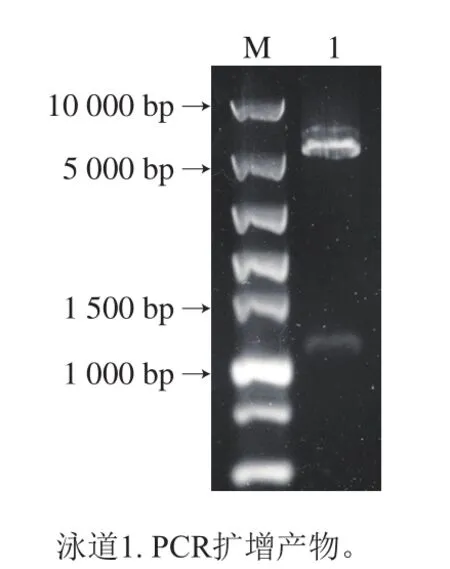
图2 pET28a-S.cerevisiae PDE1基因重组质粒基因酶切后DNA电泳结果Fig.2 Electrophoresis of recombinant plasmid of pET28a-S.cerevisiae PDE1 after enzymatic digestion
目的基因酶切产物与质粒酶切产物连接成重组pET28a-S.cerevisiae PDE1质粒,并进行DNA电泳,酶切后胶回收。如图2所示,重组质粒酶切后出现2 条带,其中6 000 bp处是载体pET28a的基因条带,1 000 bp处与S.cerevisiaePDE1目的基因预期片段大小一致。对重组质粒进行核苷酸测序鉴定,结果表明重组质粒的插入序列与PDE1目的基因一致,证明S.cerevisiae PDE1基因重组质粒载体构建成功。
2.2 重组pET28a-S.cerevisiae PDE1质粒的诱导表达
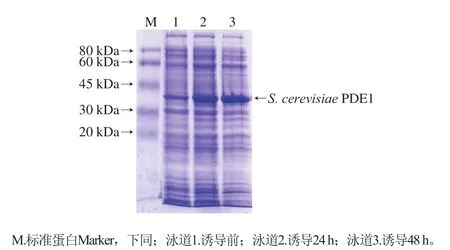
图3 S.cerevisiae PDE1蛋白菌体不同时间取样SDS-PAGEFig.3 SDS-PAGE electrophoresis of S.cerevisiae PDE1 protein at different time points of induction
15 ℃诱导培养48 h后,共收获蛋白菌体5.5 g/L。在诱导前、诱导24 h和48 h后分别取样,进行SDS-PAGE。由图3可知,随着诱导时间的延长,目的蛋白表达量随之提高,分子质量在30~45 kDa之间的蛋白含量增加迅速,这与S.cerevisiaePDE1蛋白的预期分子质量(约40 kDa)相符。该结果表明,在该诱导条件下能够获得大量的S.cerevisiaePDE1蛋白。
2.3 重组蛋白的分离纯化
2.3.1 菌体破碎与Ni-NAT柱分离纯化
为判断S.cerevisiaePDE1蛋白的水溶性和结合Ni-NAT柱的能力,确保后期能够更好地进行分离纯化。对菌体破碎后总蛋白、离心后上清蛋白和沉淀蛋白分别取样,进行SDS-PAGE,结果如图4a所示。S.cerevisiaePDE1表达菌体细胞破碎后,上清液中蛋白含量比较少,表明该蛋白诱导表达主要为包涵体。然而上清液表达蛋白结合Ni-NAT柱的能力较好。如图4b所示,上清液表达蛋白和Ni-NAT柱结合时目的条带在总条带中比例达到70%以上,说明该上清液蛋白能通过Ni-NAT柱亲和层析进行初步分离纯化,且产率较高。经过Ni-NAT柱的纯化,杂蛋白含量明显减少,其中最主要的杂蛋白分子质量约为60 kDa。
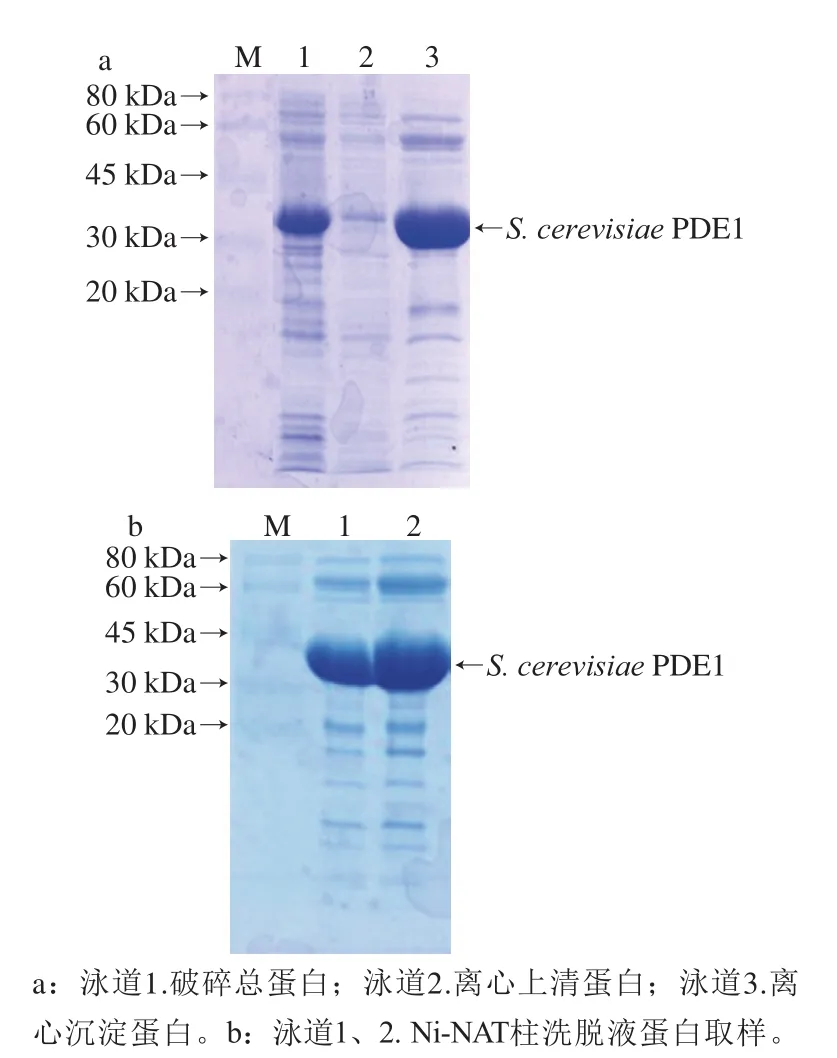
图4 S.cerevisiae PDE1细胞破碎取样(a)及Ni-NAT柱纯化取样(b)SDS-PAGEFig.4 SDS-PAGE analysis of S.cerevisiae PDE1 from disrupted cells (a)and its purified product by nickel affinity column chromatography (b)
2.3.2 蛋白酶切及Q-Sepharose柱纯化
为切除S.cerevisiaePDE1蛋白的His-Tag标签,使其能够更好地与Q-Sepharose柱结合,对Ni-NAT柱纯化后的蛋白进行酶切处理,而后利用Q-Sepharose系统进行第2次纯化。对Q-Sepharose系统纯化的蛋白取样进行SDSPAGE分析,结果如图5所示。能看出经过Q-Sepharose系统纯化蛋白的纯度增加,杂蛋白条带减少,但是E.coli蛋白条带依旧未完全除去。
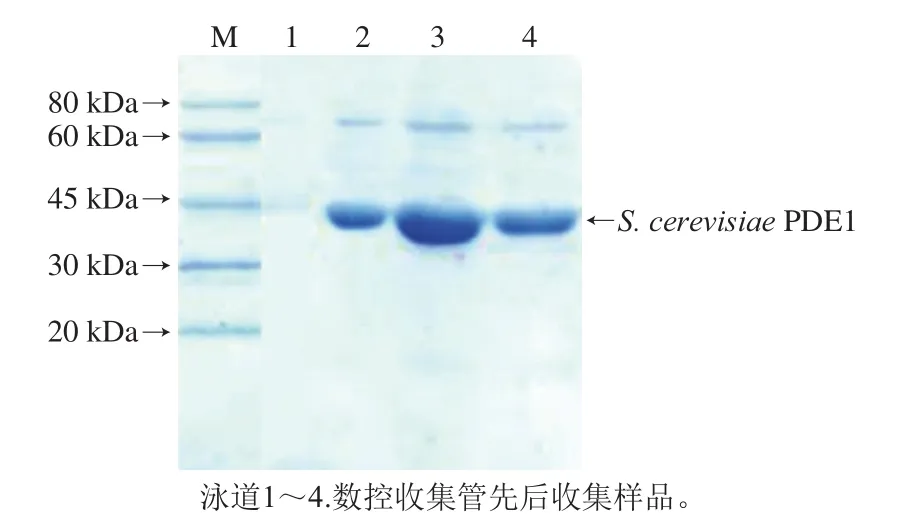
图5 S.cerevisiae PDE1蛋白过Q-Sepharose柱纯化后SDS-PAGEFig.5 SDS-PAGE analysis of S.cerevisiae PDE1 protein after purification by Q-Sepharose column chromatography
2.3.3 蛋白浓缩及Sephacryl S200柱纯化
经Q-Sepharose柱纯化的蛋白离心浓缩至11 mL,上样至Sephacryl S200柱纯化。取样SDS-PAGE结果见图6a,经过Sephacryl S200柱的纯化,E.coli蛋白条带已基本去除,证明经过Sephacryl S200分子筛的纯化,目的蛋白的纯度得到了进一步的提高。将纯化后的蛋白浓缩取样,进行非变性凝胶电泳,结果见图6b,目的蛋白能够进入非变性凝胶,未出现凝聚现象,且几乎不含多聚体蛋白。至此,经过3 次纯化后,获得了高纯度的目的蛋白,且能够保持其相应的结构及生物活性。
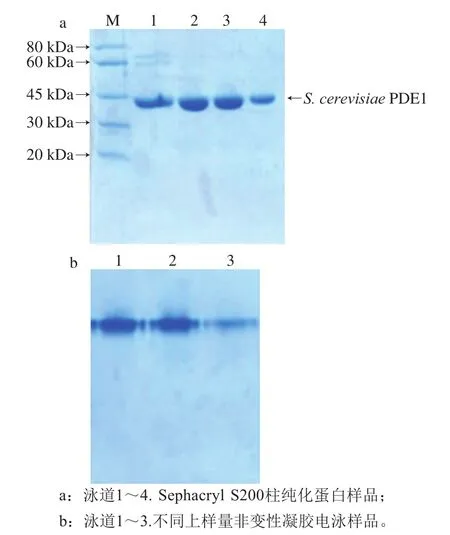
图6 S.cerevisiae PDE1蛋白Sephacryl S200柱纯化SDS-PAGE(a)及非变性凝胶电泳(b)结果Fig.6 SDS-PAGE (a) and non-denaturing gel electrophoresis (b) of S.cerevisiae PDE1 protein after purification by S200 column chromatography
2.3.4S.cerevisiaePDE1蛋白酶活性分析
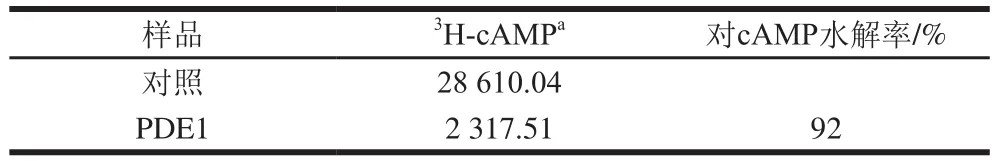
表1 S.cerevisiae PDE1蛋白原液酶活性Table 1 Enzymatic activity of S.cerevisiae PDE1
对纯化后的PDE1蛋白进行酶活性测定,如表1所示,PDE1蛋白活性较高,对cAMP的水解率到90%以上。进一步证明经3 次纯化后重组目的蛋白能够保持其原有的生物学特性。为验证S.cerevisiaePDE1蛋白是一种能够同时水解cAMP和cGMP的酶,通过不同质量浓度的蛋白水解两种底物的产物转化率进行线性分析,结果如图7所示。随着蛋白质量浓度的升高,底物转化率也升高,且具有一定的线性关系。结果证明,在MnCl2催化下,150 ng/mLS.cerevisiaePDE1蛋白对3H-cAMP的底物转化率能达到50%左右,而在300 ng/mLS.cerevisiaePDE1蛋白质量浓度条件下,对3H-cGMP的底物转化率能达到50.5%。此结果印证了关于S.cerevisiaePDE1蛋白是一种双底物酶的结论,同时,也证明本实验纯化蛋白具有很高的生物活性。
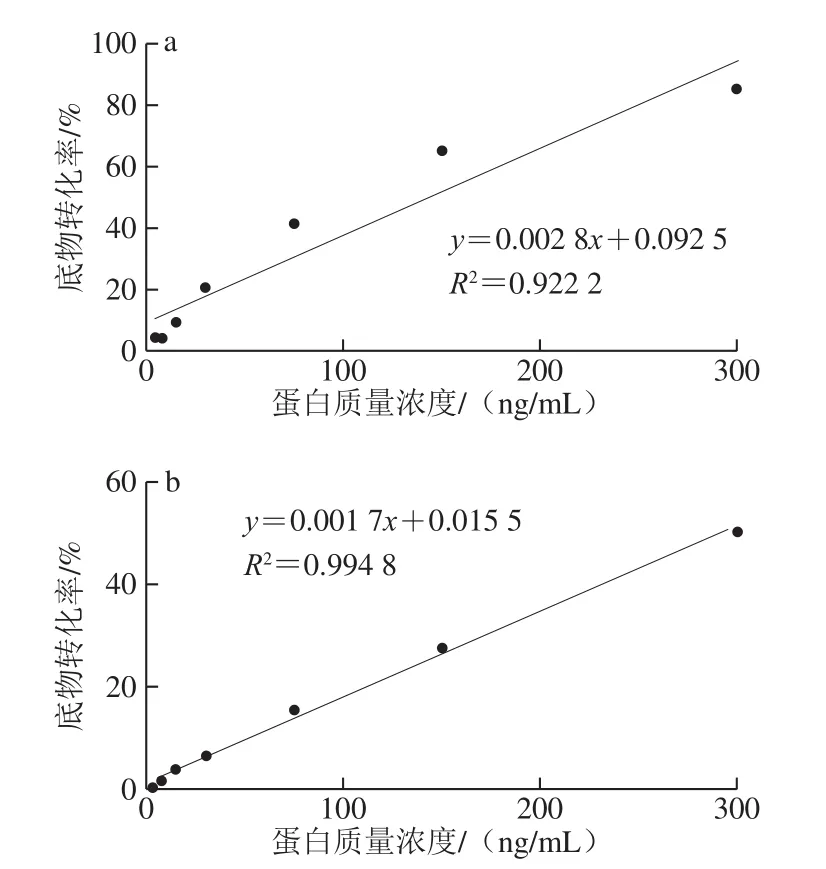
图7 MnCl2催化条件下S.cerevisiae PDE1蛋白对3H-cAMP(a)和3H-cGMP(b)的底物转化率Fig.7 Substrate conversion efficiency of 3H-cAMP (a) and 3H-cGMP (b)by different concentrations of S.cerevisiae PDE1 under MnCl2 catalysis
3 讨 论
由于在蛋白诱导表达的过程中,E.coli自身蛋白表达以及折叠机制缺乏等原因,将会产生大量的杂蛋白[26]。为了提高目的蛋白的产率,增加蛋白纯度,本研究使用最优的诱导条件,获得大量的目的蛋白表达菌体。运用多种分离纯化系统对表达蛋白进行纯化,目的是制备具有高酶活性和高纯度的重组S.cerevisiaePDE1蛋白。研究发现,多数关于重组蛋白的纯化仅利用重组蛋白的His-Tag进行Ni柱亲和系统纯化,如陈慧芹等[27]利用Ni柱纯化羊口疮病毒蛋白ORFV035,制造多克隆抗体;余璐璐等[28]利用Ni柱纯化拟南芥氰丙氨酸合酶CYS-C1,并利用该纯化蛋白制备多克隆抗体。通过对以往研究的总结,发现用作结构生物学研究的蛋白一般会使用2 种以上的纯化系统进行纯化。如吴忧[29]利用Ni柱亲和层析和分子筛对人源IDO1蛋白进行纯化,得到95%以上纯度的蛋白质,并与小分子物质进行共晶实验研究;郭团玉等[30]利用阴离子交换柱与分子筛对酵母细胞中表达的丝氨酸蛋白酶进行纯化,最后制备获得无活性突变体晶体;张兰君等[31]通过Ni柱纯化以及凝胶亲和层析对人源Pelota C端结构域进行充分纯化,从而获得该结构域的晶体结构。目前,有部分关于S.cerevisiaePDE1的功能及调控机制的研究,如蒲毅楠[32]对S.cerevisiaePDE1在细胞内的定位与内部调控机制进行了研究,认为PDE1趋向于集中到细胞生长最为茂盛的部位调节细胞生长和分化;马琳等[15]对PDEs突变株S.cerevisiae发酵产生的果酒抗氧化性和挥发物质差异性进行研究,PDEs突变株相比野生菌株具有更高的发酵速度,且Δpde2/Δpde2菌株具有更高的抗氧化能力。这些研究为S.cerevisiaePDE1在酵母细胞内的分子调控起着重要的作用。而本研究所制备的大量高纯度S.cerevisiaePDE1蛋白具有极高的生物活性,能够大量水解cAMP,利用该纯化蛋白开展下一步结构生物学的研究,有望通过该研究为胞外调控S.cerevisiae细胞的生长代谢打下基础。
猜你喜欢
自然灾害学报(2022年2期)2022-05-10
当代水产(2022年1期)2022-04-26
食品与发酵工业(2021年7期)2021-04-27
中国学校体育(2021年10期)2021-04-26
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
科技资讯(2018年16期)2018-10-26
科技资讯(2017年12期)2017-06-09
中国调味品(2017年2期)2017-03-20
中国光学(2015年1期)2015-06-06
