
1例多次复发骶尾部畸胎瘤病例报告及文献回顾
2020-09-16 01:15胡邦邹齐张迪唐业电任东林
结直肠肛门外科 2020年4期
胡邦,邹齐,张迪,唐业电,任东林△
1中山大学附属第六医院肛肠外科 广东广州510655
2德昌县中医医院肛肠外科 四川凉山州615500
骶尾部畸胎瘤(sacrococcygeal teratoma,SCT)是一种发生于骶前间隙或骶尾部区域的肿瘤,包含来自两种及以上原始生殖细胞类型。骶尾部畸胎瘤主要见于新生儿,平均每4万名新生儿中就有1名患病,男女比例约为1:3[1]。大多数骶尾部畸胎瘤可在产前诊断中发现,50%~70%在出生后数天内发现,约10%在2岁后发现,成人骶尾部畸胎瘤极为罕见[2]。因为骶尾部畸胎瘤非常罕见,不仅是儿科医师和产科医师,甚至儿外科医师很少接触到此类病例,在临床工作中,亦尚未有骶尾部畸胎瘤的诊治规范。
本文报告1例反复手术(14次手术)的骶尾部畸胎瘤患者的诊治经过,并通过分析相关文献探讨该疾病的诊断和治疗要点,供临床参考。
1 临床资料
患者女性,39岁,门诊以主诉“出生后骶尾部反复肿痛伴溢脓30余年,14次手术后”收入院。患者出生后即发现骶尾部长有一直径约1 cm×1 cm的包块,出生后3个月在当地医院行第1次局部肿物切除手术,术后切口未愈合,且局部肿块逐步生长至直径约3 cm;出生后10个月时再次在当地医院行肿物局部切除手术,术后切口仍未愈伴溢脓;至患者12岁,骶尾部肿物已增长至突出体表10 cm以上,伴有局部肿痛不适,遂在当地上级医院分期(2次手术)切除局部肿物并引流脓肿,切除骶尾部肿块重约15公斤,术后切口仍一直未愈合伴骶尾部反复流脓,期间未予医学治疗;至患者22岁,以“骨结核”在某结核病防治中心治疗2年半,病情无好转;至患者35岁,局部感染加重,在某骨科医院拟“骨髓炎”请国外专家会诊行介入手术治疗,术后症状无任何改善,转至另一家医院以“肛瘘”行局部清创或脓肿引流手术8次,术后局部感染有改善但仍存在切口不愈合;至患者38岁,患者骶尾部局部感染加重,伴有阴道流脓渗液,在当地某医院行脓肿切开引流,并行病理活检提示为畸胎瘤(病史见表1),为进一步治疗来我院就诊。
入院体格检查:左侧卧位骶尾部见广泛不规则手术瘢痕,瘢痕区域可见局部皮肤破溃流脓伴红肿(见图1),触痛明显;肛门阴道指诊:阴道距肛缘2 cm可及一直径0.3 cm凹陷,按压见脓性液体溢出,直肠黏膜光滑,肠壁质软,退出指套无染血。实验室辅助检查:血液分析示WBC 11.48×109/L,N%74.6%;肿瘤相关标志物CEA 1.73 ng/mL,AFP 2 U/mL。影像及内镜辅助检查:肠镜所见回肠末段及结直肠黏膜未见异常;盆腔MRI显示骶前、双侧坐骨肛门窝、臀部多发纤维瘢痕及脓肿、窦道形成,考虑囊肿合并感染(见图2),脓腔与下段阴道后壁相通考虑阴道瘘形成。胸腹盆CT排除肝、肺及腹腔内转移病灶,骶尾部病灶考虑囊肿合并感染。术前我院对外院病理活检会诊意见提示畸胎瘤。
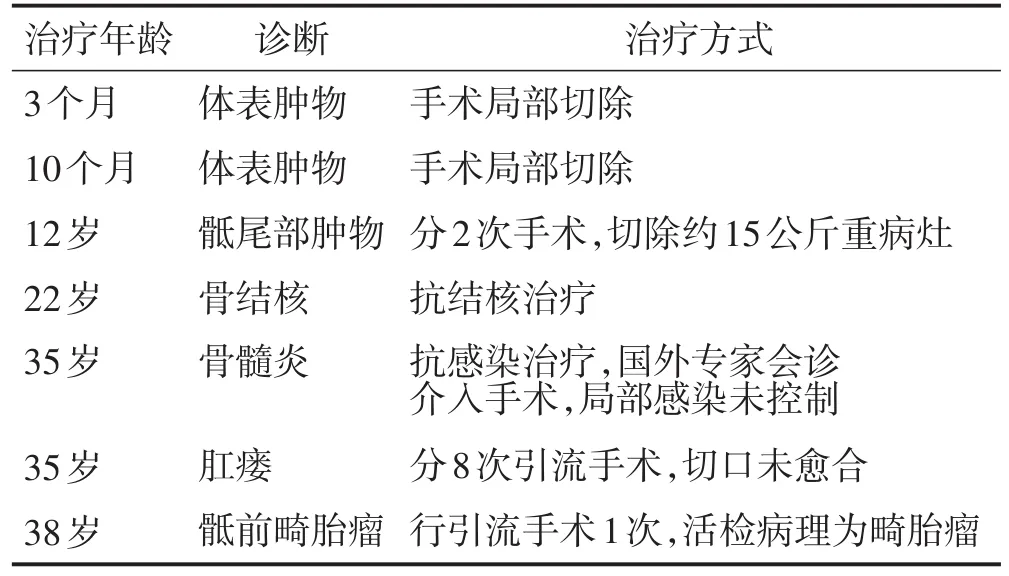
表1 患者诊治经过
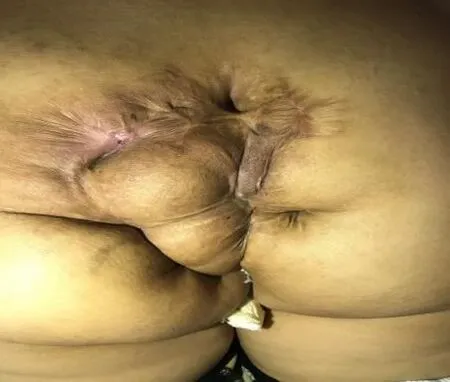
图1 术前骶尾部外观
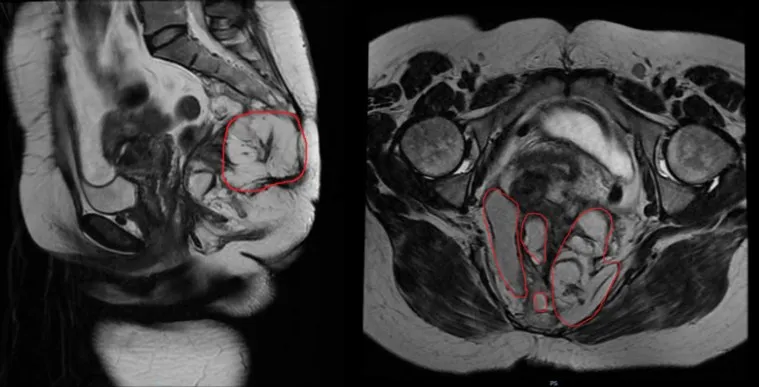
图2 术前盆腔MRI所见病灶(红色标记区域)
2 治疗
2.1 手术方案
结合患者诊治病史、典型盆腔MRI表现和病理活检结果,诊断为骶尾部畸胎瘤,经多学科会诊后考虑畸胎瘤为良性可能性大,局部感染后形成阴道瘘,病灶与尾骨关系密切,考虑采用经骶尾入路切除病灶及尾骨,阴道瘘口旷置引流,骶尾部切口一期缝合。
患者取折刀位,气管插管全身麻醉后常规消毒铺巾,采用经骶入路梭形切口切除原有手术瘢痕。逐层切开皮肤、皮下组织,见肿瘤主体部位于骶前和直肠左后方,与直肠和肛提肌以及左侧闭孔内肌关系密切,术中切除尾骨充分显露出肿瘤。为保证肿瘤完整切除,分离边界为囊壁和正常组织(包括肛门括约肌),切除病灶已累及的肛提肌进入肛提肌上间隙,继续锐性结合钝性分离肿瘤和直肠的间隙以及肿瘤和左侧闭孔内肌的间隙,完整切除肿瘤(见图3)。检查创面情况(见图4),检查直肠壁未见破损,经肛门充气试验阴性,重建肛提肌后逐层关闭切口(见图5)。
2.2 术后处理
患者术后禁食1周,卧床2周,术后每日予甲硝唑200 mL经引流管灌洗切口,切口予持续负压吸引。术后第14天拆线,引流管于术后1个月拔除,切口一期愈合。术后病理提示:成熟性囊性畸胎瘤(见图6)。
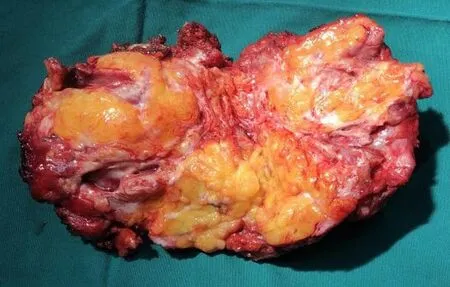
图3 切除肿物标本
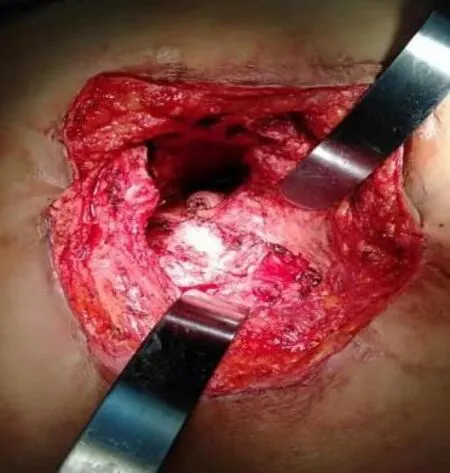
图4 手术创面
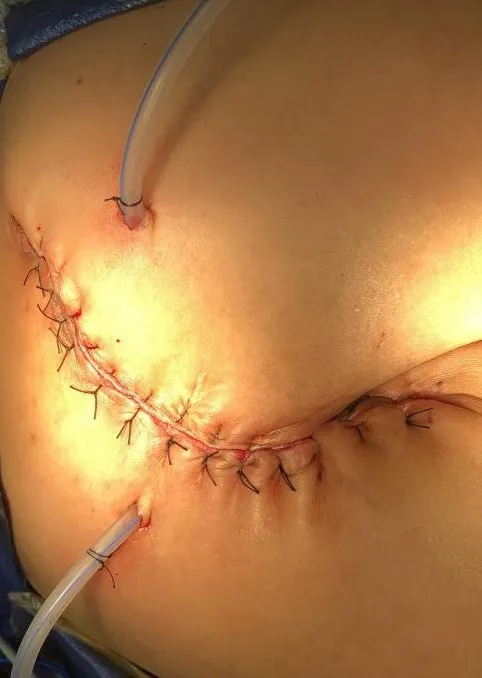
图5 术毕切口外观

图6 术后病理图片(HE,×40)
2.3 随访结果
患者术后1个月返院复查,切口正常愈合(见图7)。自诉肛门控便功能正常,Wenxer失禁评分0分;自诉阴道无粪液及气体排出,阴道指诊原阴道瘘口已愈合。
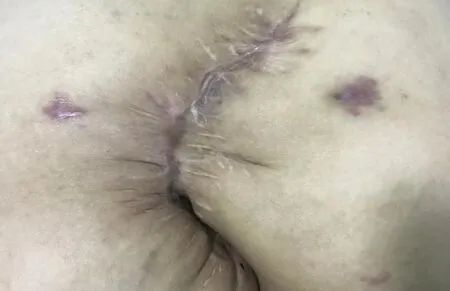
图7 术后 1个月切口外观
3 讨论
骶尾部畸胎瘤最早由Taguchi[3]于1869年描述,由于其复杂性被称为“巨大肿瘤”。其发生的原因是在人类胚胎发育过程中,由多个原始胚层细胞产生的一种先天性胚胎肿瘤。骶尾部畸胎瘤由骨、毛、牙等复杂成分组成,在病理学上可分为成熟畸胎瘤和未成熟畸胎瘤。90%的新生儿骶尾部畸胎瘤从患者体表可见,根据骶尾部畸胎瘤的发生部位,通常采用Altman分类法进行分类[4](见图8),包括以下类型:Ⅰ型,肿瘤多由突出于盆腔外病灶构成;Ⅱ型虽然盆腔内有病灶,但盆腔外病灶较大;Ⅲ型虽然盆腔外有病灶突出,但盆腔及腹部病灶较大;Ⅳ型盆腔外无病灶,肿瘤仅由盆腔和腹部组成。Ⅳ型SCT少见,发病率约为10%,症状出现晚,肿瘤发现晚,预后差。
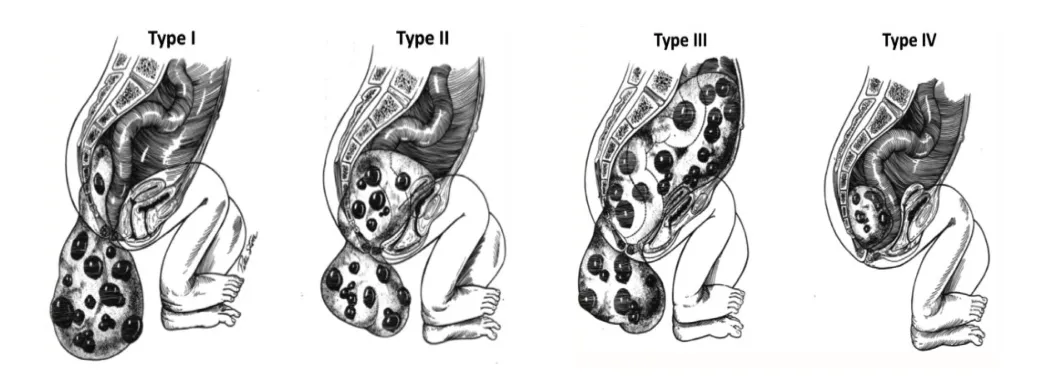
图8 骶尾部畸胎瘤的Altman分类(图片引用自参考文献[4])
尽管大多数婴儿骶尾部畸胎瘤本质上是良性肿瘤,但也有一些发展成巨大的肿瘤,导致大量出血、高输出量心力衰竭和弥散性血管内凝血(DIC),新生儿期也可能出现致命结局[5];随着年龄的增长,畸胎瘤会有恶变倾向,畸胎瘤的感染发生率为20%~30%,恶变率为1%~12%[6],血清甲胎蛋白(AFP)水平可作为监测畸胎瘤恶性转化及治疗效果的参考指标。因此,骶尾部畸胎瘤早期诊断和治疗对预后至关重要。本例患者出生后发现骶尾部肿物,受限于当时的医疗水平和接诊医师对该病的认识不足错过了治疗的最佳时机,并给后续的诊断和治疗带来了错误的指向,导致了该患者反复十余次的手术及术后复发,并因为反复的感染导致了阴道瘘。所幸是该患者最终得到了彻底的治愈,同时完整地保留了阴道和肛门器官,术后病理也未见有恶变的病理成分。
在成人中,骶尾部畸胎瘤更常表现为盆腔囊肿而无任何临床症状,因此很难被发现,发现时部分已经出现癌变,预后较新生儿骶尾部畸胎瘤差。本例患者出生后即存在盆腔外病灶,但在成年才确诊,期间多次不恰当的诊治与成人SCT本身非常罕见、绝大多数医师几乎没有诊治经验有关。直到2017年,日本骶尾部畸胎瘤研究项目委员会制定了可供临床医师参考的目前唯一的SCT临床诊疗指南[5]。
病史和临床症状是骶尾部畸胎瘤诊断的主要依据,对于临床症状不典型的病例,B超、盆腔CT和MRI均是有效的辅助检查方法。其中,MRI评估肿瘤的特征有一定的优势,包括大小、形态、囊内容物、滋养血管,以及肿瘤与周围器官的关系[7]。骶尾部畸胎瘤的良恶性鉴别缺乏典型的影像学特征,局部浸润和局部淋巴结肿大可能提示潜在的恶性[1]。不推荐对骶尾部畸胎瘤疑似病例常规行术前病理活检[8],但因本例患者有十余次的手术史并接受过各种治疗,致使囊肿的完整性被破坏且合并有感染性的脓肿和瘘管,需行术前病理活检明确诊断从而制定更完善的治疗方案。
对于骶尾部畸胎瘤的治疗,无论良性还是恶性均需要手术完整切除才有可能治愈,而肿瘤类型和侵袭范围决定了手术方案和术后处理。在Altman Ⅰ型病例中,适合创伤较小的骶尾入路手术,而经腹的手术适用于腹盆部病灶较大的患者。主体部延伸至S3以上的骶尾部畸胎瘤需要经腹入路手术[9],而主体部位于S3以下的骶尾部畸胎瘤手术则采用经骶尾入路切除[10],对于经腹或经骶尾入路不能完全切除的巨大骶尾部畸胎瘤,则采用前后联合入路手术[11]。根据笔者所在团队的经验,如果病灶主要是囊性的,经骶尾入路的手术则不必局限于病灶主体部在S3以下。肿瘤经骶尾切口暴露后,可以吸出囊液,使肿瘤塌陷,然后完全切除病灶。对于巨大的骶尾部畸胎瘤,术中出血的风险较高,骶中动脉或髂内动脉供血血管的预结扎是有效的[5]。对于SCT手术中是否切除尾骨尚存在争议[12],切除尾骨的主要原因包括:(1)对于一个较大的病灶,术中切除尾骨可以更好地暴露手术野;(2)骶尾部畸胎瘤可能来源于尾骨的多能干细胞;(3)骶尾部畸胎瘤常常紧贴尾骨,切除尾骨可以有效防止复发。本例患者因为反复十余次的手术治疗,致使局部病灶的边界不清,病变与尾骨、直肠、肛门括约肌、闭孔内肌、肛提肌以及阴道关系密切,手术的难点在于完整切除病灶,术中将尾骨和部分受累的肛提肌与病灶一起完整切除,同时尽可能保留肛门功能及相关器官的完整性。
受限于该病的罕见性,目前全球仅有日本制定了SCT的诊疗指南,且该指南引用的临床研究中尚无随机对照研究,证据等级偏低。SCT虽然临床发病率低,但我国总人口基数大,结直肠肛门外科临床中接触到此类病例的机会越来越多,如何早期诊断并合理治疗SCT需要结直肠外科医师更多地关注该病,以期制定适合我国患者的诊疗指南。
猜你喜欢
舰船科学技术(2022年20期)2022-11-28
家庭科学·新健康(2021年1期)2021-03-31
保健与生活(2021年6期)2021-03-16
家庭科学·新健康(2020年12期)2020-02-04
红领巾·探索(2019年6期)2019-08-01
保健与生活(2019年1期)2019-01-13
保健与生活(2018年5期)2018-04-28
家庭百事通·健康一点通(2018年1期)2018-01-25
中国男科学杂志(2016年5期)2016-12-01
中国塑料(2015年4期)2015-10-14
