
通络止痛胶囊中4 种成分测定及HPLC 指纹图谱建立
2020-09-15 03:17谭翔匀
中成药 2020年8期
周 和 谭翔匀 冯 华 王 奥
(遵义市产品质量检验检测院, 贵州遵义563002)
通络止痛胶囊由虎杖、当归、独活、威灵仙、全蝎、蕲蛇、杜仲等16 味药材组成,方中虎杖利湿退黄、清热解毒、散瘀止痛;当归补血活血、调经止痛;独活祛风除湿、通痹止痛,配合其他药材可充分发挥该制剂搜风通络、活血止痛、祛风除湿的功效,主要用于骨痹、风湿痹血瘀阻络症所致肌肉麻木及关节、腰颈疼痛等症[1]。但该制剂药味多,成分复杂,传统的单一成分测定难以全面控制其质量,而且存在费时费力、成本高昂的缺点,以及对照品质量不稳定、供应不充足等因素的制约。
近年来,一测多评法越来越受到重视,在2015 年版《中国药典》 编制大纲一部纲要中,国家药典委员会也重点提出要引入该方法[2],它在仅使用1 种对照品的情况下,利用相对校正因子来同时测定其他成分的含有量,具有经济、高效、制约因素少的优势,已成功应用于清热止咳颗粒、细辛、新疆软紫草、罗布麻叶等多种中药材及其复方制剂的质量控制[3⁃10]。另外,指纹图谱可反映整体化学成分信息,而一测多评法可用于定量,两者联合应用能更科学地评价中药及其复方制剂的整体质量[11⁃17]。因此,本实验建立一测多评法[18]同时测定通络止痛胶囊中虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯的含有量,并结合HPLC 指纹图谱来反映该制剂整体化学成分信息,可为其质量控制提供参考。
1 材料
1.1 仪器 LC⁃1260 型高效液相色谱仪(美国Ag⁃ilent 公司);AB204⁃S 型电子天平(北京中恒日鑫科技有限公司);KQ⁃500DV 型数控超声波清洗机(昆山市超声仪器有限公司)。
1.2 试剂与药物 虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯对照品(中国食品药品检定研究院,批号分别为11575⁃200502、111773⁃201614、110822⁃201710、111583⁃201605)。通络止痛胶囊(批号161201、161202、161203 购于遵义市中医院,批号19091601、19091602、19091603、19092301、19092302、19092303、19092304,自制)。甲醇、磷酸为色谱纯(天津市科密欧化学试剂有限公司);其他试剂均为分析纯;水为纯净水(杭州娃哈哈集团有限公司)。
2 方法与结果
2.1 色谱条件 Waters XSelect C18色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇(A) ⁃0.5% 磷酸(B),梯度洗脱(0~15 min,80%~60% B;15~25 min,60%~50% B;25~35 min,50% B;35~45 min,50%~30%B;45~60 min,30%B);体积流量1.0 mL/min;柱温25 ℃;检测波长330 nm;进样量10 μL。
2.2 溶液制备
2.2.1 对照品溶液 精密称取各对照品适量,甲醇溶解并稀释至刻度,摇匀,制成分别含虎杖苷0.830 4 mg/mL、阿魏酸0.053 6 mg/mL、蛇床子素0.782 9 mg/mL、二氢欧山芹醇当归酸酯1.245 5 mg/mL 的贮备液,精密吸取0.2、0.5、1.0、2.0、4.0 mL,置于5 个10 mL 量瓶中,甲醇稀释,即得。
2.2.2 供试品溶液 取胶囊内容物适量,研细,取约3.0 g,精密称定,置于具塞锥形瓶中,精密加入30 mL 甲醇,称定质量,超声处理60 min,静置至室温,甲醇补足减失的质量,摇匀,取续滤液,0.22 μm 微孔滤膜过滤,即得。
2.2.3 阴性样品溶液 按处方比例制备缺独活(含蛇床子素、二氢欧山芹醇)、缺虎杖(含虎杖苷)、缺当归 (含阿魏酸) 的阴性样品,按“2.2.2” 项下方法制备,即得。
2.3 方法学考察
2.3.1 系统适应性考察 取对照品、供试品、阴性样品溶液,在“2.1” 项色谱条件下进样测定,结果见图1。由此可知,各成分色谱峰达到基线分离,理论塔板数均大于5 000,与相邻色谱峰的分离度大于1.5,阴性无干扰。
2.3.2 线性关系考察 取“2.2.1” 项下对照品溶液,在“2.1” 项色谱条件下进样10 μL 测定,以溶液质量浓度为横坐标(X),峰面积为纵坐标(Y) 进行回归,分别以信噪比(S/N) 3、10 计算检测限、定量限,结果见表1,可知各成分在各自范围内线性关系良好。
2.3.3 精密度试验 取同一供试品溶液(批号161201),在“2.1” 项色谱条件下进样测定6 次,测得虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯峰面积RSD 分别为0.39%、1.04%、0.30%、0.81%,表明仪器精密度良好。
2.3.4 重复性试验 取胶囊(批号161201) 内容物适量,按“2.2.2” 项下方法平行制备6 份供试品溶液,在“2.1” 项色谱条件下进样测定,测得虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯峰面积RSD 分别为0.45%、1.70%、0.57%、0.94%,表明该方法重复性良好。

图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.3.5 稳定性试验 取同一供试品溶液(批号161201),于0、2、4、6、8、10、12、24 h 在“2.1” 项色谱条件下进样测定,测得虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯峰面积RSD 分别为0.93%、1.86%、1.35%、1.62%,表明供试品溶液在室温下24 h 内稳定性良好。
2.3.6 加样回收率试验 精密称取各成分含有量已知的胶囊9 份,每份1.50 g,每3 份1 组,精密加入80%、100%、120% 水平对照品溶液,按“2.2.2” 项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定,计算回收率,结果见表2。
2.3.7 相对校正因子计算 取“2.2.1” 项下对照品溶液,在 “2.1” 项色谱条件下进样2、5、10、12、15、20 μL 测定,以虎杖苷为内标,计算其他3 种成分的相对校正因子,结果见表3。
2.4 耐用性考察
2.4.1 仪器、色谱柱 本实验考察了Agilent 1260、Waters 2695 色谱仪,以及XSelect C18、Eclipse C18、Diamonsil C18色谱柱对相对校正因子的影响,结果见表4,可知均无明显影响(RSD<2.50%)。
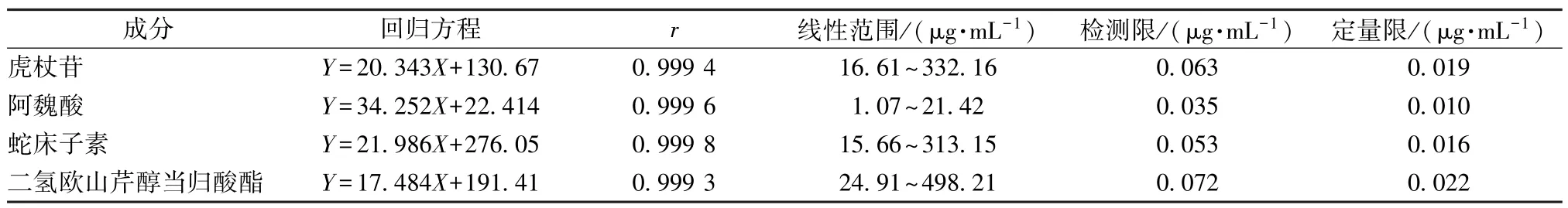
表1 各成分线性关系Tab.1 Linear relationships of various constituents
2.4.2 检测波长 本实验考察了检测波长332、330、328 nm 对相对校正因子的影响。结果,阿魏酸相对校正因子分别为0.552、0.553、0.556,RSD 为0.38%;蛇床子素相对校正因子分别为0.835、0.832、0.835,RSD 为0.21%;二氢欧山芹醇当归酸酯相对校正因子分别为1.153、1.146、1.151,RSD 为0.31%,可知均无明显影响。
2.4.3 体积流量 本实验考察了体积流量0.8、1.0、1.2 mL/min 对相对校正因子的影响。结果,阿魏酸相对校正因子分别为 0.558、0.553、0.568,RSD 为1.36%;蛇床子素相对校正因子分别为0.837、0.832、0.825,RSD 为0.73%;二氢欧山芹醇当归酸酯相对校正因子分别为1.159、1.146、1.165,RSD 为0.84%,可知均无明显影响。
2.4.4 柱温 本实验考察了柱温20、25、30 ℃对相对校正因子的影响。结果,阿魏酸相对校正因子分别为0.547、0.553、0.560,RSD 为1.18%;蛇床子素相对校正因子分别为 0.821、0.832、0.835,RSD 为0.89%;二氢欧山芹醇当归酸酯相对校正因子分别为1.158、1.146、1.132,RSD 为1.14%,可知均无明显影响。
2.4.5 磷酸体积分数 本实验考察了磷酸体积分数0.4%、0.5%、0.6% 对相对校正因子的影响。结果,阿魏酸相对校正因子分别为0.537、0.553、0.562,RSD 为2.30%;蛇床子素相对校正因子分别为0.861、0.832、0.839,RSD 为1.79%;二氢欧山芹醇当归酸酯相对校正因子分别为1.105、1.146、1.143,RSD 为2.02%,可知均无明显影响。
2.5 色谱峰定位 精密吸取对照品溶液,在“2.1” 项色谱条件下进样测定,结果见表5。由此可知,不同仪器、色谱柱对相对保留时间的影响较小,RSD 均小于5%,表明相对保留时间法可用于定位色谱峰。

表2 各成分加样回收率试验结果(n=9)Tab.2 Results of recovery tests for various constituents (n=9)

表3 各成分相对校正因子Tab.3 Relative correction factors of various constituents
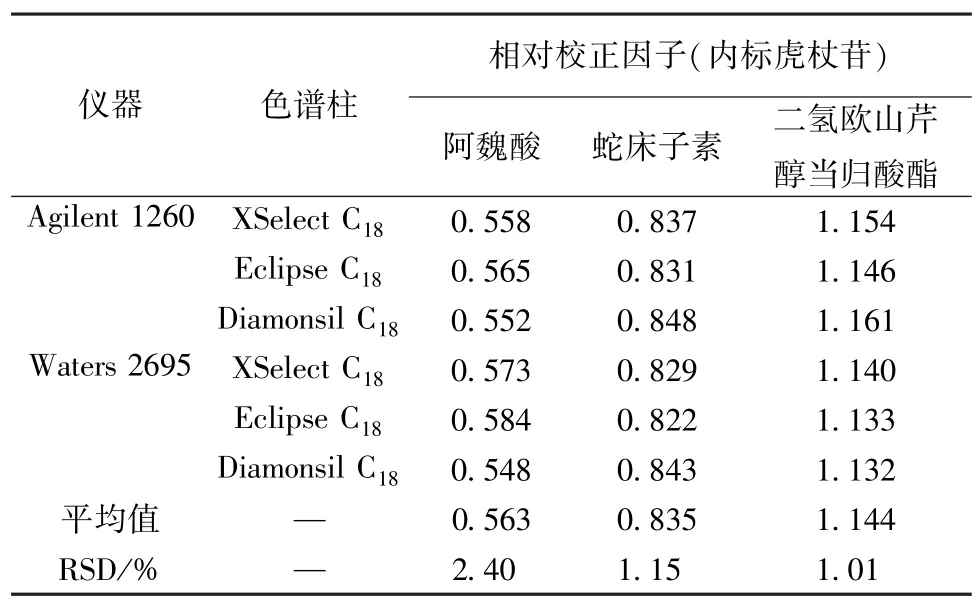
表4 不同仪器、色谱柱对相对校正因子的影响Tab.4 Effects of different instruments and columns on relative correction factors
2.6 样品含有量测定 取10 批胶囊,按“2.2.2”项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定,计算含有量,结果见表6,可知2 种方法所得结果接近,差异百分比 (PD) 均小于2%。

表5 各成分相对保留时间Tab.5 Relative retention time of various constituents
2.7 HPLC 指纹图谱建立
2.7.1 精密度试验 取同一供试品溶液(S1,批号161201),在“2.1” 项色谱条件下进样测定6次,以虎杖苷为参照峰,测得各共有峰相对保留时间RSD 为0.15%~0.46%,相对峰面积RSD 为0.12%~0.97%,所采集指纹图谱的相似度均大于0.97,表明仪器精密度良好。
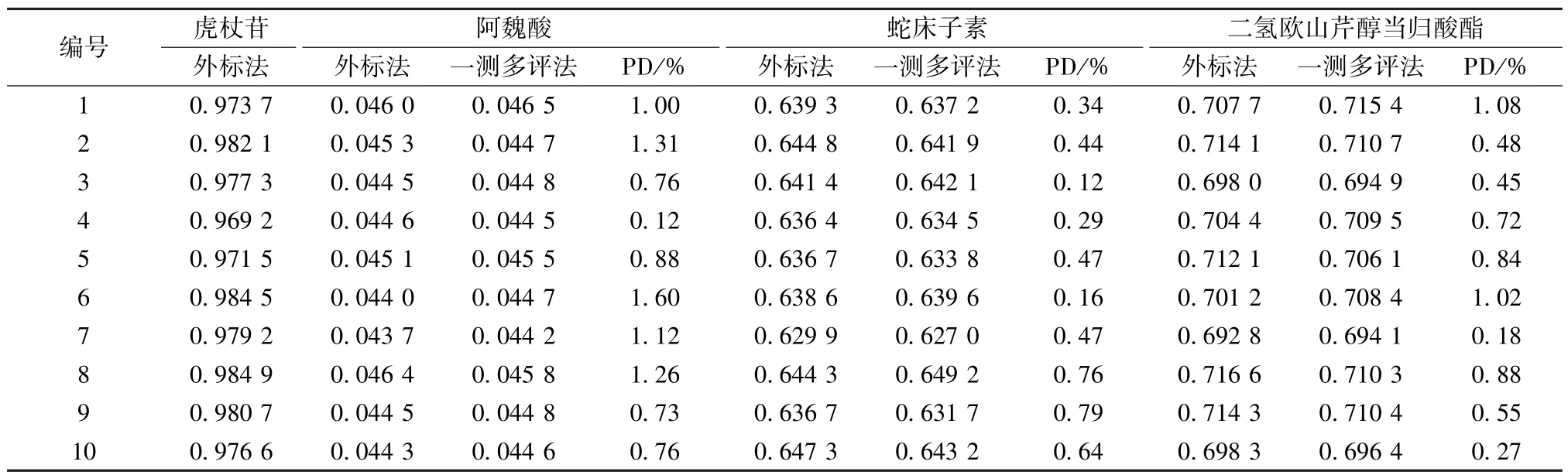
表6 各成分含有量测定结果(mg/g)Tab.6 Results of content determination of various constituents (mg/g)
2.7.2 稳定性试验 取供试品溶液(S1),于0、2、4、8、12、16、24 h 在“2.1” 项下色谱条件进样测定,以虎杖苷为参照峰,测得各共有峰相对保留时间、相对峰面积RSD 均小于1.72%,指纹图谱相似度均大于0.96,表明溶液在室温下24 h内稳定性良好。
2.7.3 重复性试验 取样品(S1),平行制备6份供试品溶液,在“2.1” 项色谱条件下进样测定,以虎杖苷为参照峰,测得各共有峰相对保留时间RSD 均小于0.59%,相对峰面积RSD 均小于1.43%,指纹图谱相似度均大于0.96,表明该方法重复性良好。
2.7.4 指纹图谱生成及相似度评价 取10 批胶囊内容物适量,按“2.2.2” 项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定,采用中药色谱指纹图谱相似度评价系统(2012 版),选择样品S1 图谱作为对照指纹图谱,通过多点校正全谱匹配后确定21 个共有峰,生成指纹图谱共有模式,见图2。另外,10 批样品相似度分别为0.984、0.996、0.973、0.980、0.982、0.962、0.971、0.978、0.985、0.987,均大于0.95,表明不同批次样品之间的差异较小,稳定性良好。
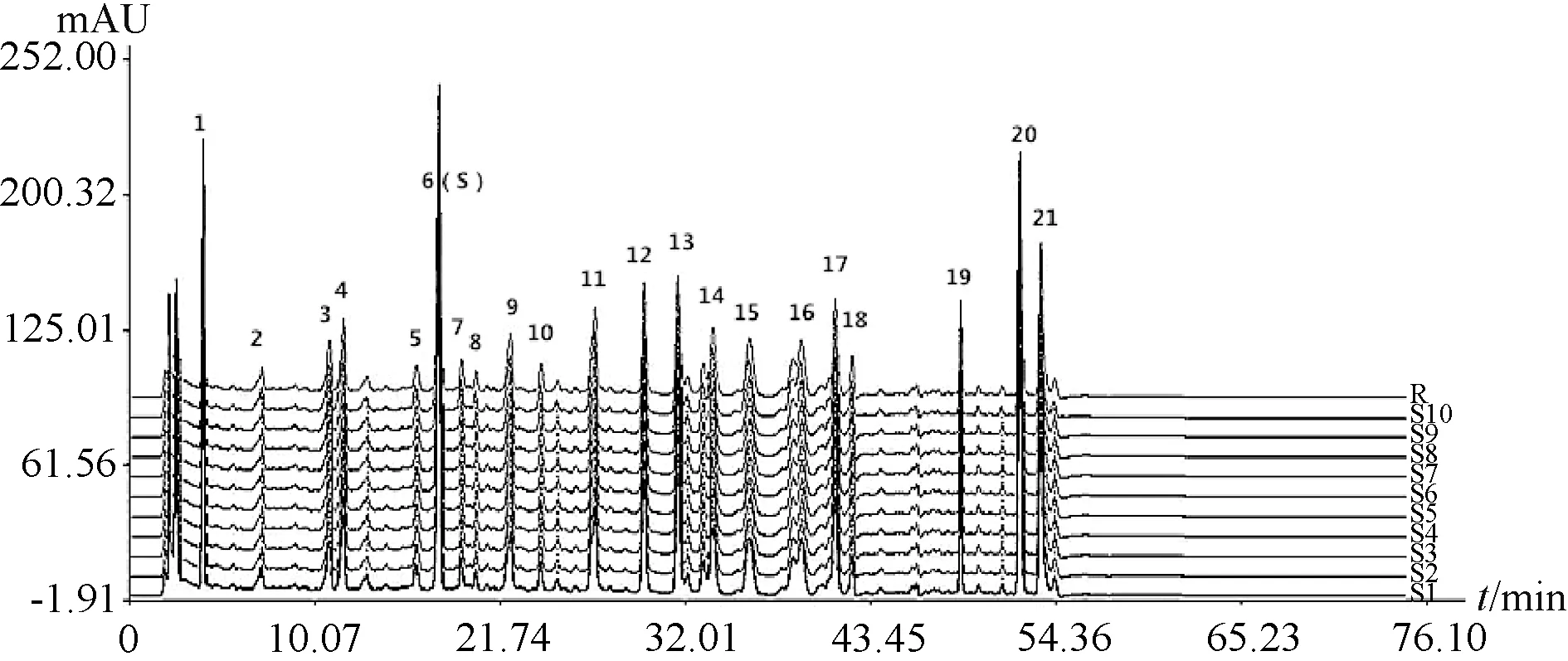
图2 10 批样品HPLC 指纹图谱Fig.2 HPLC fingerprints for ten batches of samples
2.7.5 共有峰 21 个共有峰的峰面积总和占色谱峰总面积的93.51%,表明它代表性较好,然后在供试品溶液中加入对照品溶液以考察相应共有峰是否增高,同时与对照品溶液图谱进行比较(图1),确定6 号峰为虎杖苷,7 号峰为阿魏酸,20 号峰为蛇床子素,21 号峰为二氢欧山芹醇当归酸酯,并选择峰形好、位置适中、前后无色谱峰干扰的虎杖苷作为参照峰(S 峰)。再计算各共有峰相对保留时间、相对峰面积RSD,结果见表7~8,可知它们均小于2.0%,表明指纹图谱稳定性良好。
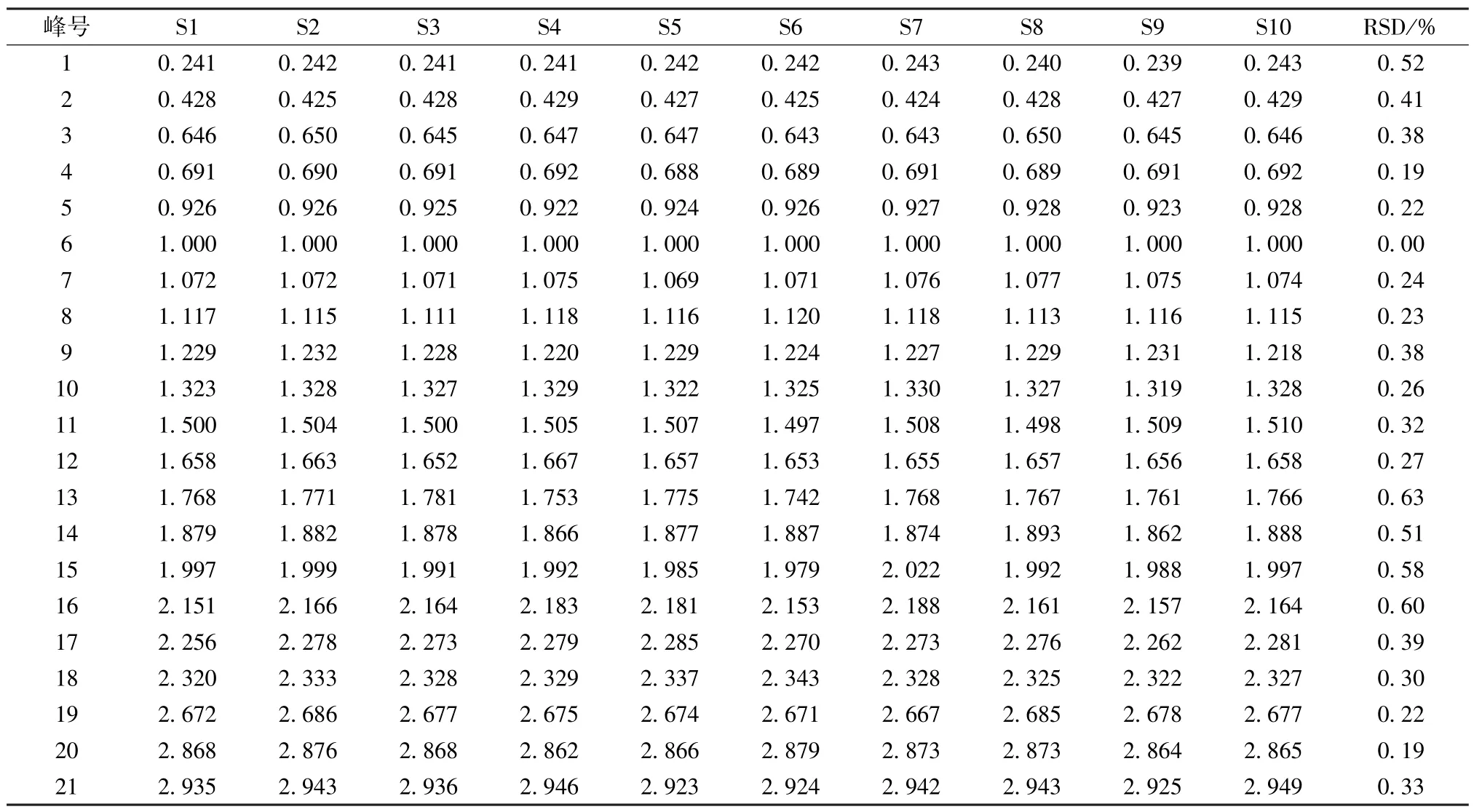
表7 共有峰相对保留时间Tab.7 Relative retention time of common peaks
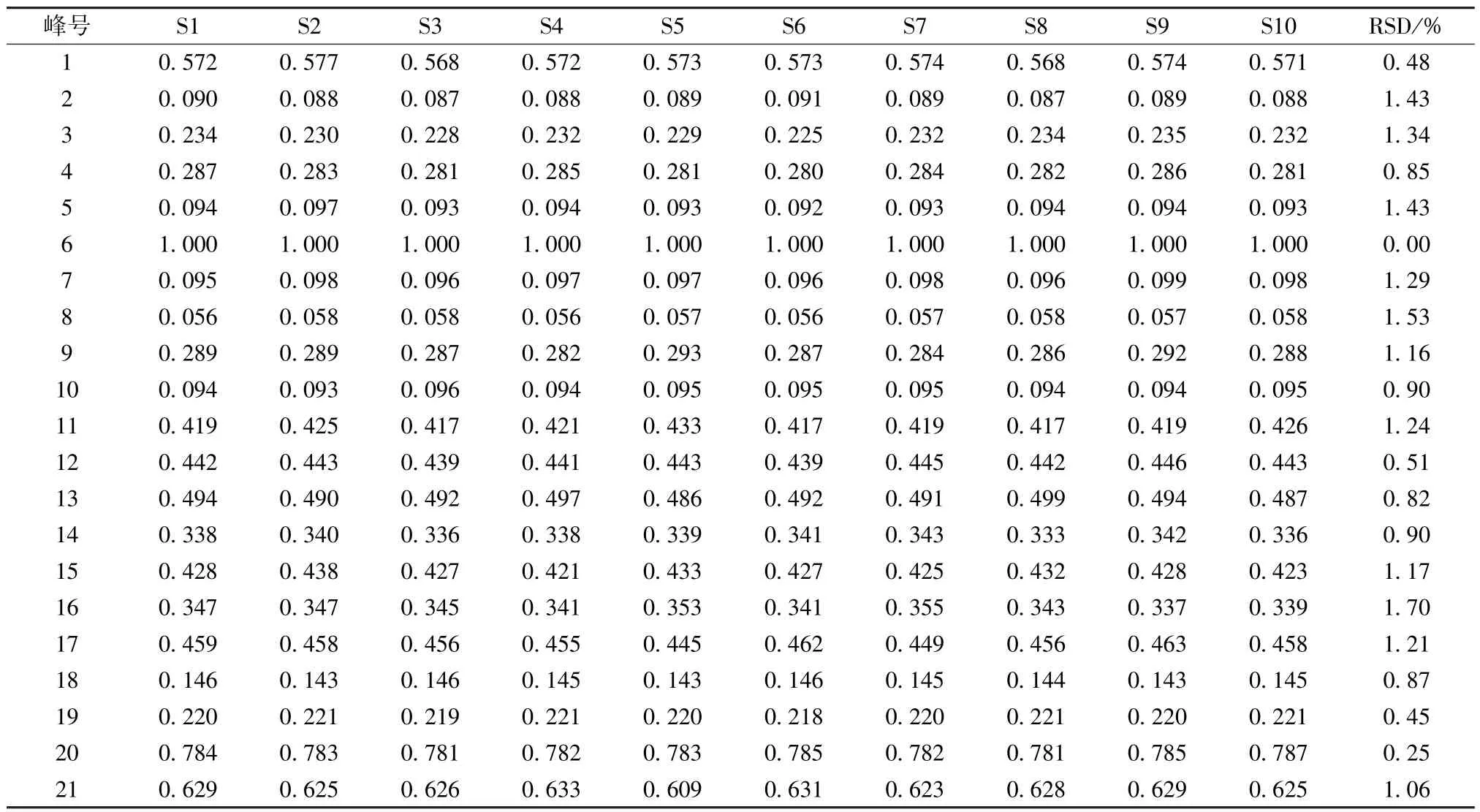
表8 共有峰相对峰面积Tab.8 Relative peak areas of common peaks
3 讨论
3.1 指标成分、内标选择 根据通络止痛胶囊中16 味药材的主要成分及其药理作用,本实验考察了虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯、毛蕊花糖苷、蒿本内酯、松脂醇二葡萄糖苷。其中,毛蕊花糖苷、蒿本内酯含有量太低,松脂醇二葡萄糖苷不易提取分离,故选择虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯作为指标成分。另外,虎杖苷在供试品溶液中含有量高,位置适中,峰形好,分离度大,故选择其作为内标。
3.2 前处理方法选择 本实验比较了乙醇、甲醇,发现后者提取率高,基线平稳,图谱指纹性强,故选择甲醇作为提取溶剂;比较了超声提取、回流提取、索氏提取,发现三者无明显差异,但超声提取操作方便快捷,易于制备供试品溶液,故选择其作为提取方法;比较了提取时间50、60、70 min,发现提取60 min 后各成分提取率基本一致,故选择60 min 作为提取时间。
3.3 色谱条件选择 本实验所测成分的极性相差较大,故采用梯度洗脱模式以实现快速分离。考察了甲醇⁃水、甲醇⁃磷酸、甲醇⁃醋酸、乙腈⁃水、乙腈⁃磷酸、乙腈⁃醋酸,发现甲醇-0.5%磷酸洗脱效果最好,故以其为流动相。各成分紫外吸收波长分别为虎杖苷306 nm、阿魏酸316 nm、蛇床子素330 nm、二氢欧山芹醇当归酸酯330 nm,四者比较接近,采用DAD 检测器进行190~400 nm 全波长扫描,通过分析三维图谱,兼顾信号强度和检测灵敏度,发现在330 nm 波长附近各成分色谱图的基线噪音较低,均有较高的响应,有利于含有量测定,故选择其作为检测波长。
4 结论
指纹图谱具有整体性、相似性的特点,可快速判断药味是否缺少,但不能准确反映化学成分信息;一测多评法定量准确,但受制于中药的复杂性,也并非所有活性成分均能如此测定。本实验首次建立通络止痛胶囊HPLC 指纹图谱,并通过一测多评法同时测定虎杖苷、阿魏酸、蛇床子素、二氢欧山芹醇当归酸酯的含有量,发现所得结果与外标法接近,而且该方法简便高效,重复性好,可为该制剂整体质量控制提供参考。
猜你喜欢
中国野生植物资源(2022年1期)2022-02-15
神经药理学报(2021年4期)2021-01-05
今日中国·西班牙文版(2020年7期)2020-07-04
国学(2020年1期)2020-06-29
保健与生活(2020年12期)2020-06-23
现代园艺(2020年11期)2020-06-13
家庭影院技术(2018年11期)2019-01-21
摄影之友(影像视觉)(2017年10期)2017-11-07
摄影之友(影像视觉)(2017年1期)2017-07-18
家庭医学(2017年5期)2017-06-02
