
金属卤化物钙钛矿光催化材料研究进展
2020-09-14 03:32赵韦人
发光学报 2020年9期
黄 浩 赵韦人 李 杨 罗 莉
(广东工业大学 物理与光电工程学院, 广东 广州 510006)
1 引 言
随着世界人口的日益增长及社会经济的飞速发展,传统的化石能源消耗给地球生态环境造成了巨大压力。 气候变暖、环境污染正日益明显地影响人类社会的可持续发展。 因此,大力发展可持续的清洁能源显得尤为迫切。 太阳能是一种清洁、可持续的能源,其每天辐射到地球表面的能量是全人类一年消耗总能量的200 倍。 因此,合理高效地利用太阳能是解决目前面临的环境、能源问题的最理想途径。 1972 年,Fujishima 和Honda首次报告了利用光能进行TiO2水解析氢实验[1],开启了光催化应用的研究热潮。 通过光催化反应将太阳能转化为化学能,是高效利用太阳能的最具代表性的一种策略[2]。 至今已报道包括氧化物钙钛矿[3-6]、金属氧化物[7-9]、硫化物[10]、氮化物[11]、磷化物[12]以及非金属材料[13]等许多光催化材料。 但目前光催化材料普遍面临的问题是:光吸收范围窄、可见光利用率低、制备工艺复杂、载流子利用率及光催化效率低等[14-16]。 因此,寻找开发新型高效、稳定的光催化材料仍十分迫切。
近年来,以铅卤钙钛矿材料(Lead halide perovskites,LHPs)为代表的金属卤化物钙钛矿材料(Metal halide perovskites,MHPs)以其简易的制备方法和优异的光电特性,如光致发光量子产率高[17-19]、带隙可调[20]、光吸收系数高[21]、载流子扩散长度大[22]和寿命长[23]等,在太阳能电池[24]、发光二极管[25]、激光器[26]、光电探测器[27]等光电领域中得到了广泛研究。 特别地,在短短十年间MHPs 太阳能电池的功率转换效率从起初的3.8%[24]发展到25.2%[28],并有望在未来突破30%[29]。 MHPs 优异的光电特性及其在太阳能电池领域的飞速发展,使其成为一种极具发展前景的新型高效光催化材料[30-31]。
钙钛矿的化学分子式为CaTiO3,最早于1839年被Gustav Rose 发现,随后俄罗斯矿物学家L.A. Perovski(1792—1856)命名了该种化合物[32]。此后,将具有与CaTiO3相似晶体结构的材料统称为钙钛矿材料。 其中,氧化物钙钛矿材料通常由二价金属阳离子(如Ca2+、Sr2+、Ba2+等)、四价金属阳离子(如Ge4+、Ti4+、Zr4+等)和氧离子组成,结构通式为ABO3;金属卤化物钙钛矿的结构通式为ABX3,其阳离子A通常为一价金属离子或一价有机离子(如Cs+、Rb+、CH3NH3+(MA+)、CH(NH2)2+(FA+)等)、阳离子B为二价金属离子(如Pb2+、Sn2+、Cu2+、Ge2+等)或三价阳离子(如Bi3+、Sb3+等),阴离子X为卤素离子(Cl-、Br-、I-)。 当A为有机阳离子时,称为有机-无机杂化卤化物钙钛矿材料,如MAPbX3、FAPbX3、MASnX3等;当A为金属阳离子时,称为全无机卤化物钙钛矿材料,如CsPbX3、CsSnX3等;当阳离子B为除Pb2+以外的其他阳离子时,称为无铅卤化物钙钛矿材料,如MASnX3、MA3Bi2X9等。
本文从MHPs 的结构特点出发,概述了光催化反应的作用机理以及MHPs 在光催化应用中的优势,分析讨论了MHPs 的环境稳定性并对提高MHPs 光催化反应稳定性的方法进行了概括总结,综述了近年来MHPs 在光催化应用中的发展现状,包括光催化析氢、光催化CO2还原、光催化有机转化等,对比分析了不同光催化策略的作用机理及发展中面临的困难与挑战,最后分析和展望了高效稳定金属卤化物钙钛矿光催化材料的发展前景。
2 金属卤化物钙钛矿的结构
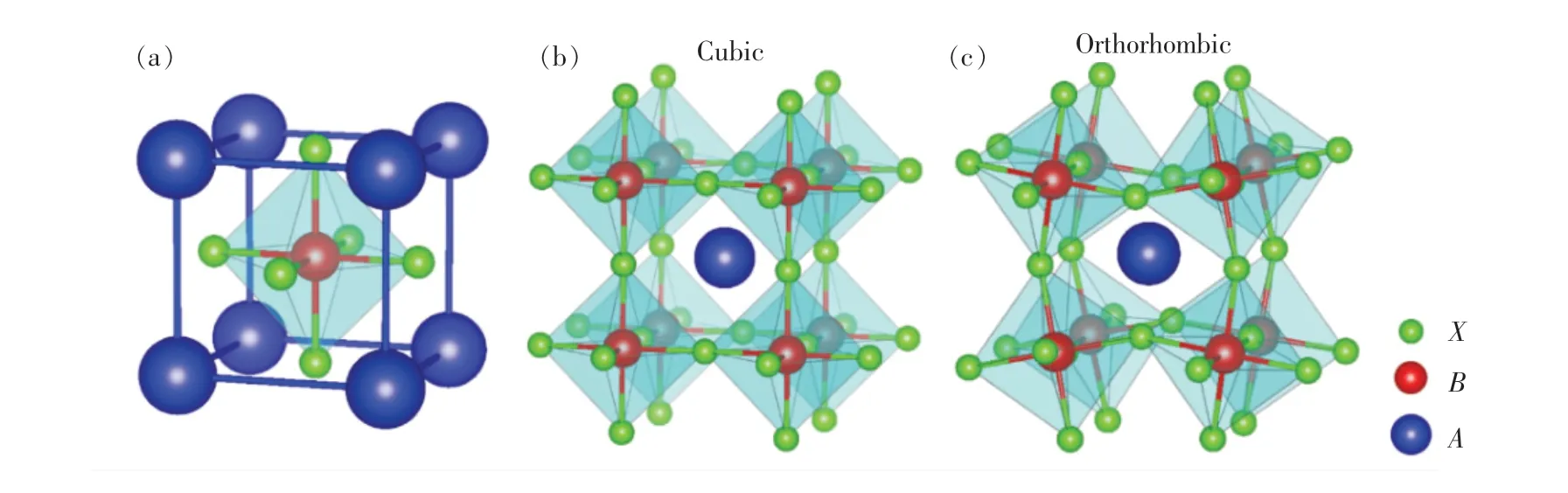
图1 (a)MHPs 的立方相结构示意图;(b)MHPs 的三维立方相结构;(c)MHPs 的正交相结构。Fig.1 (a)Depiction of metal halide perovskites with cubic structure. Three-dimension crystal structure of metal halide perovskites with cubic(b) and orthorhombic(c) phase.
理想MHPs 的晶体结构为具有高度对称性的立方相结构(空间群:Pm3m),如图1(a)所示。金属阳离子A和卤素离子X(X=Cl,Br,I)分别占据立方体的顶角和面中心,6 个卤素离子X和1个阳离子B构成BX6八面体,其中阳离子B位于八面体的中央。BX6八面体通过顶角彼此相连构成了钙钛矿材料的三维框架,相邻两个八面体间的B—X—B键角成180°,阳离子A则嵌在BX6八面体形成的三维框架的空隙中,如图1(b)所示。 通常,BX6八面体间的扭曲会使得MHPs 的结构偏离立方相结构,形成对称性较低的正交相结构,如图1(c)所示。
阳离子A为12 配位离子,不同半径的阳离子A会对MHPs 的可成型性和相结构稳定造成影响:阳离子A的半径过大则无法嵌入空隙,过小则不足以支撑三维框架,使钙钛矿结构坍塌。 一般采用半经验的几何参数,Goldschmidt 容忍因子t,来预测钙钛矿材料的可成型性和结构相稳定性[33]。t可以写成:
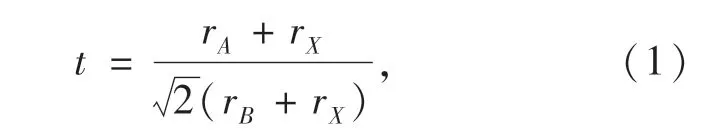
其中,rA、rB和rX分别为离子A、B和X的尺寸半径。一般认为,当t=0.813 ~1.107 时,绝大多数的钙钛矿材料可以维持其结构的稳定性[34];当t=0.9 ~1时,钙钛矿材料具有立方相结构,而在0.71 ~0.9 范围则可能形成BX6八面体扭曲的正交相;当t>1 或<0.71 时,由于BX6八面体的严重扭曲,形成非钙钛矿相结构,导致带隙加宽,电导性下降[35-36]。

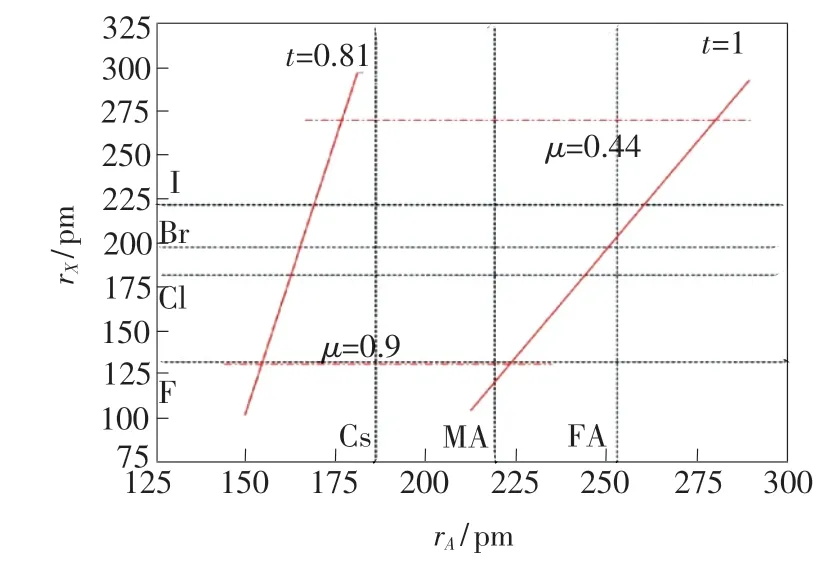
图2 LHPs 的可成型性和相稳定性关于阳离子A 和卤素阴离子X 的关系。 红色实线和虚线分别对应Goldschmidt 容忍因子t 和八面体因子μ[37]。Fig.2 Formability and phase-stability of 3D lead halide perovskites as a function of A-site cation and X-site halide anion radii. Red solid and dash represent Goldschmidt tolerance factor and octahedral factor,respectively[37].
预测钙钛矿结构相稳定性的另一个半经验几何参数为八面体因子μ:一般地,当μ=0.442 ~0.895 时,认为BX6八面体是稳定的。 根据Goldschmidt 容忍因子t和八面体因子μ,可以得到LHPs(A=Cs+,MA+,FA+)可成型性和相稳定性与离子半径(阳离子A和阴离子X)的关系,如图2 所示。 对于阳离子Cs+、MA+和FA+而言,MAPbX3的结构非常接近理想的立方相结构,而对于理想的A位阳离子,阳离子Cs+的尺寸略微偏小,阳离子FA+的尺寸则略微偏大。
需要指出的是,Goldschmidt 容忍因子t和八面体因子μ并非判断MHPs 可成型性和相结构稳定性的充分条件。 因为MHPs 为离子晶体结构,原子间的相互作用力较弱,形成能较低,因此外界因素(如温度、压力、湿度等)容易对其相结构稳定性产生较大影响。 一般地,钙钛矿材料随着温度的改变会发生结构相变,且立方相结构通常在高温下趋于稳定[38]。 例如,MAPbI3薄膜随着温度升高存在两个相变温度(160 K 和330 K),分别对应γ相向β相的转变和β相向α相的转变[39]。
3 金属卤化物钙钛矿的光催化特点
3.1 光催化机理
自然界中,绿色植物通过自然光合作用利用太阳能将CO2和H2O 转化成碳水化合物和O2,从而实现太阳能-化学能的转化。 光催化反应也称为人工光合作用,通过模拟自然光合作用将光能转化为化学能,从而实现能量转换和再利用,图3(a)、(b)分别展示了两种光合作用过程的机理[40]。 通常,光催化反应涉及3 个反应过程:(1)光捕获;(2)产生光生电子-空穴对并迁移至光催化材料表面相应的氧化-还原位点;(3)催化反应过程,光生电子和光生空穴在催化活性位点进行氧化、还原反应。 反应底物的氧化-还原电势应位于光催化材料的带隙之间,且光催化材料的导带底相对于还原电势越负,光生电子的还原效率越高,价带顶相对于氧化电势越正,光生空穴的氧化效率越高。 因此,光催化材料的光吸收系数、能级结构及光生电子-空穴对的分离和迁移对光催化反应的性能具有至关重要的影响。
3.2 金属卤化物钙钛矿的光催化优势
基于上述光催化机理的分析可知,高效的光催化材料对其能级结构、光生载流子的分离和迁移、光吸收系数等光电特性有较高的要求[41]。
MHPs 作为直接带隙半导体,其导带底主要由阳离子B的p 轨道和卤素离子X的p 轨道组成,价带顶主要由阳离子B的部分s 轨道和卤素离子X的p 轨道杂化的反键轨道组成,阳离子A则平衡三维BX6八面体框架的电负性[42]。 以MAPbX3为例,当X分别为I、Br、Cl 时,其带隙大小分别为1.6,2. 39,3. 1 eV,对应可见光的红、绿、蓝区域[43]。 通过卤素原子替换[44]或量子限域效应[45],MAPbX3的带隙可以实现整个可见光范围的覆盖。 如图3(c)所示,简易的带隙可调性使得MHPs 可以更好地与光催化反应中地氧化-还原电势相匹配。
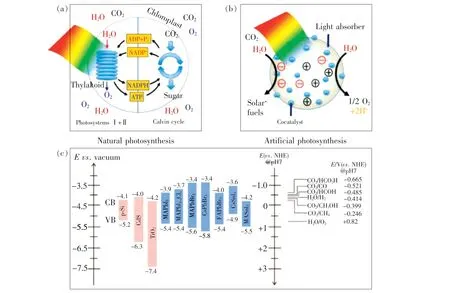
图3 (a)自然光合作用机理;(b)人工光合作用机理[40];(c)MHPs 与典型的半导体材料(p-Si、CdS、TiO2)的能级比较及反应底物(H2O、CO2)对应不同光催化产物的氧化-还原电势的排列情况[46]。Fig.3 Schematic illustration for natural(a) and artificial(b) photosynthesis[40]. (c)Conduction band and valence band potentials of representative semiconductors(p-Si, CdS, TiO2) and halide perovskites for solar-to-chemical fuel conversion[46].The relative potentials of the CO2 and water redox couples at pH=7 are plotted versus vacuum(left) and normal hydrogen electrode(NHE)(right).
目前多数光催化材料的光吸收范围局限于紫外波段,对约占太阳能40%的可见光部分吸收较少,这极大地限制了在光催化反应中对太阳能的利用率[47-50]。 由于MHPs 中B位离子的s 轨道和卤素离子的p 轨道存在的强反键耦合[51],相比于氧化物钙钛矿和氮化物钙钛矿的吸收边缘(分别为~200 nm 和~650 nm),MHPs 具有更长的吸收边缘(MAPbI3: ~700 nm)[52]。 此外,MHPs 的光吸收系数可达1 ×104~1 ×105cm-1,表现出良好的光吸收性质[21,53],因此极大地提高了其在光催化应用中对可见光的利用率。
在光催化反应中,光催化材料中的空位及表面缺陷对光生载流子的捕获会大大降低催化效率。 相较于其他半导体材料,如CdSe 和GaAs,MHPs 具有更高的缺陷容忍度[54],有效地限制了光生载流子的缺陷复合几率,提高了光生载流子的利用率。 此外,MHPs 的光生载流子寿命可达微秒量级[55-56],扩散长度可达几十到上百微米[56-57],这对光生载流子迁移到材料表面的催化活性位点提供了有力的保障。
综上所述,简易的带隙可调性、更宽的光吸收范围、更高的缺陷容忍度、较长的载流子寿命和扩散长度充分表明了MHPs 在光催化应用中巨大的发展潜力。
3.3 金属卤化物钙钛矿的环境稳定性及动态平衡概念
3.3.1 环境稳定性
光催化反应通常在连续光照射、大气环境、液相反应体系等条件下进行。 因此,MHPs 作为光催化材料需要克服环境因素(光、氧气、水分等)对其稳定性造成的影响。
以MHPs 纳米晶(Nanocrystals,NCs)为例,通常采用表面配体(油酸、油胺等)维持其单分散性和胶体稳定性。 当NCs 受到持续的光照射时,容易引起表面配体质子化,使得表面配体的吸附能力下降并从表面脱落。 表面配体脱落后导致表面缺陷态增加并引发NCs 团聚,导致光催化性能下降[58]。 此外,在氧气氛围中,光照引起的光氧化效应会使MHPs 发生分解反应[59-60]。 例如,在光照下,氧气分子和MAPbI3相互作用形成超氧自由基,随后超氧自由基进一步与MAPbI3反应使其分解为PbI2、H2O、I2和CH3NH2[59]。 相比于表面配体质子化和光氧化,水分的影响更显著。MHPs 的离子晶体结构很容易在湿度环境下发生分解[61-62]。 通过时间分辨椭圆光度法及X 射线衍射,Barnes 等揭示了MAPbI3的水解作用机理,如公式(3) ~(4)[63]:
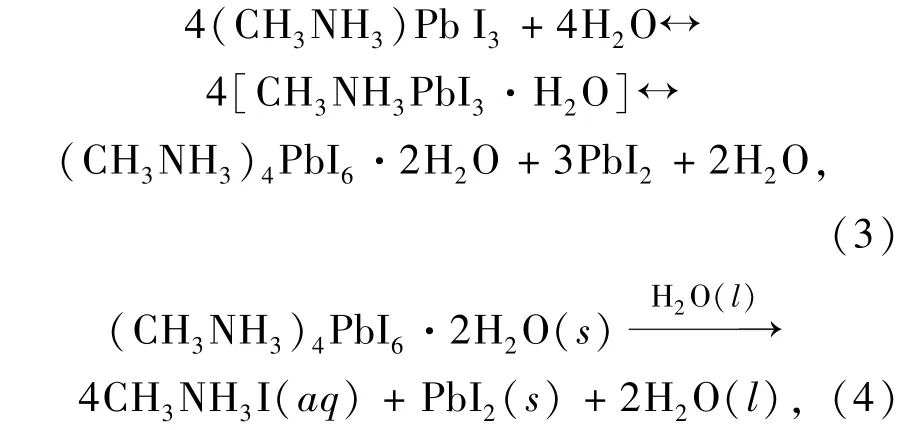
MAPbI3在水氛围中首先形成MAPbI3·H2O。 随后,MAPbI3·H2O 在H2O 的作用下形成二水化物(CH3NH3)4PbI6·2H2O 和PbI2。 因为MA+对I-的束缚力较弱,最终导致二水合物不可逆地分解为MAI、PbI2和H2O。 水分引起的分解在黑暗的条件下仍可进行,而光照会进一步加剧水分对MHPs 的分解作用。
为了提高MHPs 的环境稳定性,可以通过组分工程和表面工程两方面进行改进,如将有机官能团替换为无机阳离子[20]、调控卤素成分[21]或引入掺杂离子精确优化晶体结构[64]、对材料进行表面包裹[65]等。 此外,通过原位钝化[18]和引入等效配体[66]减少材料内部空位和表面缺陷,同样可以提升MHPs 的环境稳定性。 在光催化反应中,需要注重提升MHPs 对湿度环境的耐受性。因为光生载流子参与光催化的氧化-还原反应,因此在提升MHP 的抗水能力时也需要注重内部光生载流子向外传输的特性。 目前主要的策略包括:(1)通过MHPs 在水溶液中实现动态平衡[67];(2)优化表面配体密度,平衡其湿稳定性和光生载流子向外传输[68];(3)构建异质结构,在促进光生载流子分离和迁移的同时对MHPs 起到一定的钝化作用,常用的材料有石墨烯[69]、金属氧化物[70]、硫化物[71]、氮化物[72]以及金属有机框架材料[73]等;(4)选择非极性或弱极性的溶剂作为反应载体,降低MHPs 与水的接触,如乙酸乙酯[74]、乙腈[72]、异丙醇[75]等。
3.3.2 动态平衡概念
2016 年,Nam 等[67]首次提出动态平衡概念以解决MAPbI3在水溶液中的光催化稳定性问题,并在HI 水溶液中实现了MAPbI3光催化析氢反应。 所谓动态平衡是指当离子晶体结构的MAPbI3加入HI 水溶液时,MAPbI3分解成MA+和PbI3-,当继续添加MAPbI3时,溶液中的溶质将达到过饱和临界状态,此时MAPbI3的溶解和沉淀处于平衡状态,如图4(a)、(b)所示。 实验表明,当HI 水溶液中I-和H+的浓度满足[I-]≤[H+]、pH≤-0.5、-log[I-] ≤-0.4 时, MAPbI3可以在HI 水溶液中实现溶解-沉淀动态平衡,如图4(c)所示。 MHPs 的湿稳定性是实现高效率光催化反应的关键。 光催化析氢实验表明,通过MAPbI3在HI 水溶液中的动态平衡,MAPbI3可连续光催化析氢160 h,且催化性能并未减弱。 此外,当使用极性溶剂(二甲基甲酰胺, DMF; 二甲基亚砜,DMSO)对MAPbI3进行热处理后,MAPbI3光催化析氢速率均获得较大提升。 将Pt 作为助催化剂可实现57 μmol·g-1·h-1的析氢速率,如图4(d)所示。
随后,Goddard 等[76]通过理论模拟计算提出了基于两步铅活化-胺辅助(Pb-activated amine-assisted,PbAAA)的MAPbI3光催化析氢反应机理。计算表明,在饱和HI 水溶液中,MAPbI3光催化析氢产生的两个H 原子分别来源于两个MA+,其作用机理如图5 所示。 该反应过程由两个状态组成:(Ⅰ)首先,MA+中的一个氢原子与其邻近的Pb 原子键合,形成PbH-中间态,MA+因失去一个H 原子从而形成MA 分子,随后通过Grotthuss机制[77]重新获得一个H+形成MA+;(Ⅱ)随后,邻近的一个MA+提供一个H+与PbH-中的H-结合形成H2,而后MA+再次通过Grotthuss 机制从溶液中获得一个H+重新形成MA+。 该研究表明,MAPbI3在饱和HI 水溶液光催化析氢中不仅作为吸光材料提供光生载流子,其MA+和Pb2+对于H2的产生起到桥梁作用。
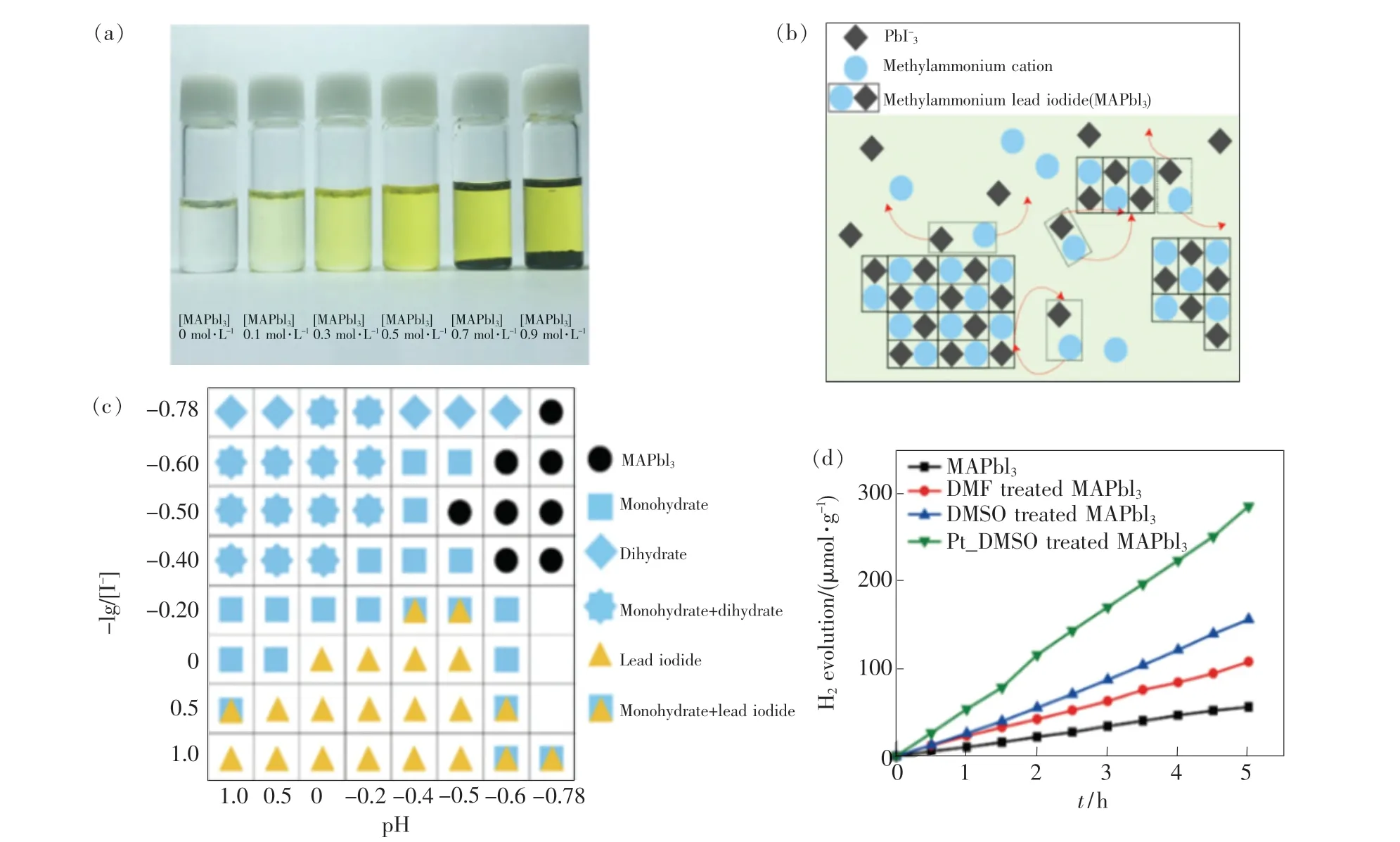
图4 (a) 不同浓度的MAPbI3 在HI 水溶液的情况; (b)MAPbI3 在饱和HI 溶液中实现溶解-沉淀动态平衡的机理;(c)不同H +和I -浓度下的结构相图;(d)MAPbI3 在不同情况下的光催化析氢性能[67]。Fig.4 (a)MAPbI3 in aqueous HI solution with different concentrations. (b)Schematic illustration of MAPbI3 powder in dynamic equilibrium with a saturated HI solution. (c)Constructed phase map as a function of [I -] and [H +]. (d)Photocatalytic H2 evolution from MAPbI3 powder under different situation[67].
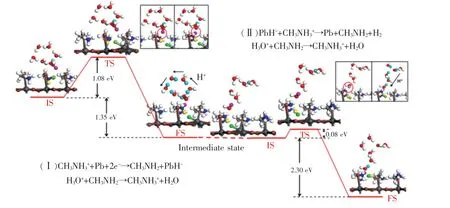
图5 两步铅活化-胺辅助的光催化析氢机理[76]Fig.5 Pb-activated amine-assisted(PbAAA) reaction pathway for H2 generation on MAPbI3 surface in acidic solvent[76]
动态平衡概念的引入对发展MHPs 在光催化领域的应用具有里程碑意义。 近几年来,MHPs光催化的应用研究得到了快速发展,下面将介绍MHPs 在光催化应用中的3 个主要分支:析氢反应、CO2还原反应、有机物转化反应。
4 金属卤化物钙钛矿在光催化中的应用
4.1 光催化析氢
典型的光催化水解析氢是吸能反应过程,反应前后体系的标准吉布斯自由能变化为+237 kJ/mol:
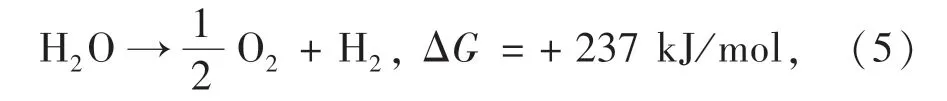
因此,光催化材料的带隙需要大于1.23 eV ( <1 000 nm),且光催化材料的导带底相对于H+/H2的还原电势(0 Vvs. NHE,pH =0)越负、价带顶相对O2/H2O 的氧化电势(1.23 Vvs. NHE,pH=0)越正时,光催化效率越高[78]。 由于水分对MHPs 结构稳定性的影响,目前MHPs 光催化析氢通常采用卤化氢水溶液(如HBr 和HI)作为反应体系,通过动态平衡的方式实现钙钛矿在湿度环境下的结构稳定。 为了提高MHPs 的光催化析氢速率,通常采用界面工程构建表面异质结构,促进光生载流子的分离和迁移效率,或采用组分工程精确调控MHPs 的光电特性以及提升晶体结构稳定性,从而获得更优的能级匹配和晶体结构。
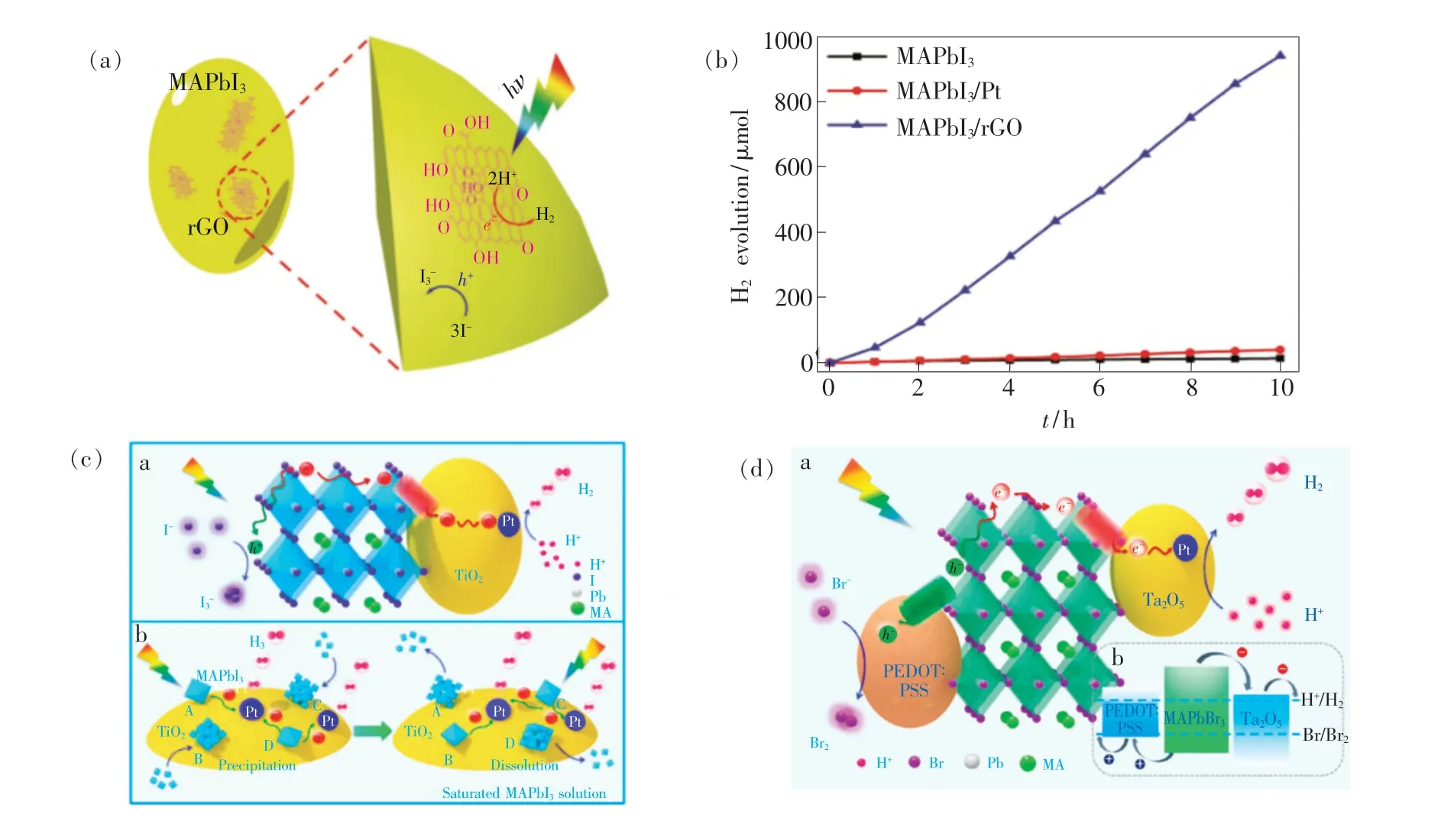
图6 (a) ~(b)MAPbI3/rGO 的光催化析氢机理及其光催化效率[69]; (c)Pt/TiO2-MAPbI3 通过纳米电荷传输通道的光催化析氢机理[70];(d)MAPbBr3/Pt-Ta2O5/PEDOT∶PSS 的光催化析氢机理及其能带结构[81]。Fig.6 (a) -(b)Schematic illustration of the H2 evolution using MAPbI3/rGO and its photocatalytic H2 evolution activities of MAPbI3, MAPbI3/Pt, and MAPbI3/rGO[69]. (c)Schematic illustration of the H2 evolution using Pt/TiO2-MAPbI3 through a nanoscale electron-transporting channel[70]. (d)Schematic illustration of the reaction mechanism for MAPbBr3 with Pt/Ta2O5 and PEDOT∶PSS as the electron- and holetransporting motifs, respectively. And schematic energy level diagrams of MAPbBr3, Ta2O5 and PEDOT∶PSS for HBr splitting reaction[81].
4.1.1 通过界面工程构造异质结构
Huang 等[69]将还原型石墨烯(rGO)与MAPbI3复合制备了MAPbI3/rGO 异质结构,将其用于饱和HI 水溶液中进行光催化析氢,光催化机理如图6(a)所示。 rGO 作为电荷接收器和传输体,可以促进光生载流子的分离与提高迁移效率,且对MAPbI3起到一定的钝化作用,使得光催化稳定性超过200 h。 得益于rGO 的复合,MAPbI3/rGO 的析氢速率达到938.9 μmol·g-1·h-1,是纯MAPbI3的67 倍,如图6(b)所示。 类似地,Zhao 等[79]将rGO与无铅钙钛矿Cs2AgBiBr6复合,使得Cs2AgBiBr6/rGO 的析氢速率相较于纯Cs2AgBiBr6提升了80倍,且光催化稳定性达到120 h。 需要注意的是,rGO 为黑色固体材料,当rGO 与MHPs 复合提高光催化速率的同时也会降低MHPs 的光吸收能力,因此实际应用中需要控制rGO 的用量以平衡光照强度与光催化速率。
考虑到rGO 会对MHPs 的光吸收带来消极影响,Zhao 等[80]制备了MA3Bi2I9/Pt,Pt 作为助催化剂提高了光生载流子的迁移效率,使得MA3Bi2I9/Pt 的析氢速率相较于纯MA3Bi2I9提高了14 倍。 此外,Li 等[70]从纳米电荷传输通道的角度来寻找合适的电荷提取材料。 因为TiO2与MAPbI3的能级相匹配,可通过TiO2在MAPbI3和助催化剂Pt 之间建立一条纳米电荷传输通道,从而显著地提升了MAPbI3与Pt 之间的电荷传输效率,作用机理如图6(c)所示。 通过调控Pt/TiO2之间的含量,Pt/TiO2-MAPbI3的析氢速率相比于同一条件下的Pt/MAPbI3提升了89 倍,达到7 300 μmol·g-1·h-1,且表观量子效率(Apparent quantum efficiency,ηAQE=Nelectron/Nphoton)高达70%(λ=450 nm),表明光生载流子得到了有效的分离和迁移。 随后,他们又进一步增加纳米电荷传输通道的数量,分别引入Pt/Ta2O5和PEDOT∶PSS 作为光生电子传输通道和光生空穴传输通道,使得光生电子和光生空穴的迁移路径分离,如图6 (d) 所示[81]。MAPbBr3/Pt-Ta2O5/PEDOT∶PSS 的光催化析氢速率较纯MAPbBr3提升了约52 倍(1 050 μmol·g-1·h-1;ηAQE=16.4,λ=450 nm)。 双纳米电荷传输通道策略进一步促进了光生载流子有序地向催化活性位点迁移,提高了光生载流子的利用率。 但由于光生空穴传输通道PEDOT∶PSS 在反应溶液中会发生团聚现象,造成电荷传输效率下降,因此仍需对该催化体系进行优化,以提高光催化反应的稳定性。
Ni3C 作为一种过渡金属碳化物常被用于光(电)催化析氢,其相比于助催化剂Pt 具有更强的电荷提取能力,可以进一步促进光生载流子的分离和迁移效率。 Tao 等[82]通过表面电荷促进的自组装方法将Ni3C 锚定在MAPbI3表面,如图7(a)所示。 MAPbI3与Ni3C 的复合使其获得了2 363 μmol·g-1·h-1的析氢速率,是纯组分MAPbI3的55 倍。 通过光致发光光谱和荧光寿命光谱发现,Ni3C 锚定在MAPbI3表面后其荧光强度几乎消失且发光寿命变短,表明Ni3C 对光生载流子的分离和迁移起到极大的促进作用,如图7(b)、(c)所示。 此外,由于Ni3C 具有较好的耐酸性,使得MAPbI3在酸性溶液中建立动态平衡时对钙钛矿结构起到保护作用,通过优化Ni3C 的用量,15% Ni3C/MAPbI3的光催化析氢稳定性达到200 h,如图7(d)所示。 类似地,Li 等[83]通过静电耦合的方法将二维黑磷(Black phosphorus,BP)锚定在MAPbI3表面,BP 促进了光催化反应中的光生载流子的利用率,使得BP/MAPbI3获得3 742 μmol·g-1·h-1的析氢速率,是纯MAPbI3的106倍。 Min 等[71]将MoS2纳米片(Nanosheets,NSs)通过原位耦合的方法制备MoS2NSs/MAPbI3,同样获得了较高的析氢速率和催化稳定性(2 061 μmol·g-1·h-1,156 h)。 通过表面耦合的方式对电荷提取材料和MHPs 进行组装,一方面,促进了MHPs 内部光生载流子的向外传输能力;另一方面,由于较强的键合力使得光催化材料获得了较高的稳定性。
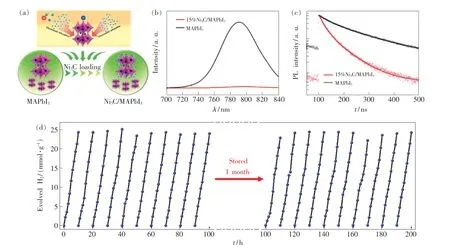
图7 (a)Ni3C/MAPbI3 的合成策略;(b)Ni3C/MAPbI3 和MAPbI3 的光致发光光谱;(c)Ni3C/MAPbI3 和MAPbI3 的荧光寿命光谱;(d)15% Ni3C/MAPbI3 的光催化稳定性[82]。Fig.7 (a)Schematic diagram of Ni3C/MAPbI3 photocatalyst preparation process. PL(b) and time-resolved PL(c) spectra based on NiC/MAPbI and MAPbI. (d)Cycling photocatalytic HER performance over 15% NiC/MAPbI[82].33333
4.1.2 通过组分工程优化光电特性和晶体结构
通过组分工程对MHPs 光电特性和晶体结构进行精确调控,实现更优的能级匹配,可以进一步提升MHPs 的光催化析氢速率。
已有研究表明,在含有两种卤素成分(Br 和I)的MHPs 中,光生载流子将从宽禁带的富Br 区域向窄禁带的富I 区域定向迁移[84-85]。 因此,调控MHPs 中卤素离子的分布,可以有效地引导光生载流子向光催化位点迁移。 例如,Huang 等采用光辅助卤素交换法分别以MAPbBr3和CsPbBr3为本体合成I-由核心到壳层浓度逐渐减小的MAPbBr3-xIx[86]和CsPbBr3-xIx[87],由于I-的梯度分布使MAPbBr3-xIx和CsPbBr3-xIx具有由核心到壳层逐渐递减的漏斗状能带结构,如图8(a)所示。 由于漏斗状的能带结构促使光生载流子向表面迁移,使得MAPbBr3-xIx/Pt 和CsPbBr3-xIx/Pt在饱和HBr/HI 水溶液中均表现出较高的光催化析氢速率,分别为2 604. 8 μmol·g-1·h-1和1 120 μmol·g-1·h-1。 但由于MHPs 的离子晶体结构性质,材料中的空位将作为卤素离子间的迁移通道[88],使得漏斗状能带结构随着卤素迁移而受到破坏,因此该催化体系的稳定性还有待进一步提升。 随后,Tao 等[89]采用Br 部分取代I 的方式制备了MAPb(I1-xBrx)3(x=0 ~0.2)。 在不使用共催化剂的情况下,MAPb(I0.9Br0.1)3的光催化析氢速率高达1 471 μmol·g-1·h-1,约是纯MAPbI3的40 倍。 密度泛函理论分析表明析氢速率的提高来源于:(1)由于Br 离子比I 离子的尺寸小,当Br 替换I 时,使得Br—Pb—Br 键的其中一个Pb—Br 键发生断裂,从而将Pb 暴露于MA+,促进了MA+中H 向Pb 迁移;(2)Pb—Br 键断裂降低了Pb—H 的能量,使得其更容易与另一个H 形成H2。 此外,较小的Br 离子提升了材料结构相的稳定性,使得MAPb(I0.9Br0.1)3的光催化析氢稳定性达到252 h。
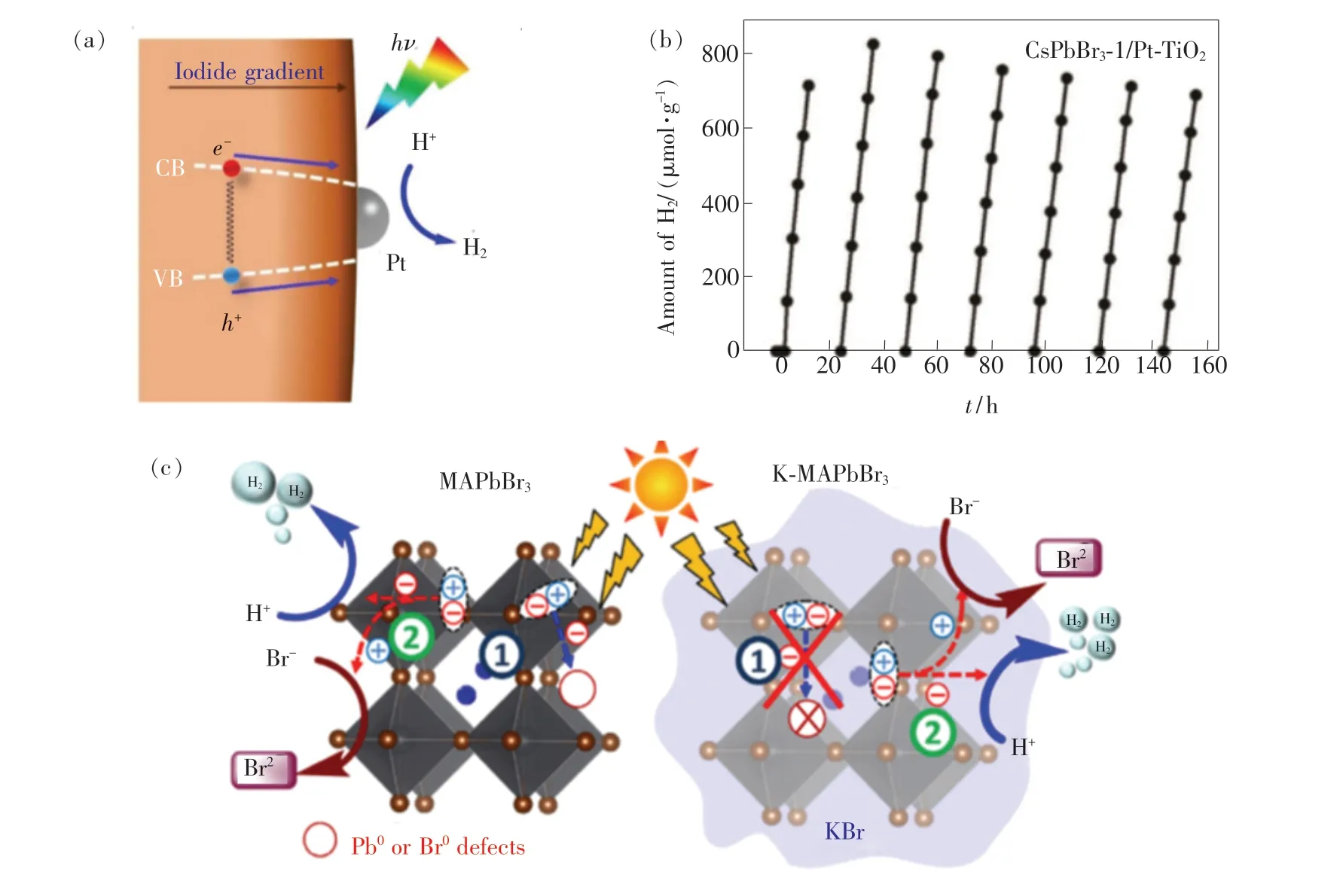
图8 (a)MAPbBr3-x Ix/Pt 的漏斗状带隙结构[86];(b)CsPbBr3/Pt-TiO2 的光催化析氢稳定性[68];(c)MAPbBr3 和KMAPbBr3 光催化析氢机理[91]。Fig.8 (a)Band gap funnel structure of MAPbBr3-xIx near the surface in MAPbBr3-xIx/Pt enhancing the photocatalytic H2 evolution on the Pt particles loaded on the surface of MAPbBr3-x Ix[86]. (b)Long-term H2 generation of CsPbBr3/Pt-TiO2 photocatalyst under visible light irradiation[68]. (c)Schematic illustration of the H2 evolution using MAPbBr3 and KMAPbBr[91].3
通过将有机官能团替换成无机离子或A位离子掺杂也是提升MHPs 稳定性的常用策略[20,90]。最近,Wang 等[68]通过优化CsPbBr3量子点(Quantum dots,QDs)表面配体密度(油酸,油胺),并采用气相光催化析氢方法,提升了CsPbBr3QDs 的湿稳定性,使其能够连续光催化160 h,如图8(b)所示。一方面,表面配体作为疏水层使得钙钛矿量子点在一定的湿度环境下稳定存在;另一方面,其高阻特性阻碍了QDs 内部光生载流子向外传输的能力。 因此,通过优化表面配体密度,可以实现MHPs 光催化析氢速率和光催化稳定性的平衡。此外,Zhao 等[91]采用K+掺杂和沉积KBr 钝化层两种策略结合的方法制备了K-MAPbBr3@ KBr,并与[Mo3S13]2-2纳米团簇组合,使得K-MAPbBr3/[Mo3S13]2-2获得了稳定的光催化析氢性能。 进一步研究表明,K+掺杂可以有效地抑制Pb0和Br0缺陷的产生,使K-MAPbBr3获得了更好的催化活性和催化稳定性,其作用机理如图8(c)所示。
不同组分的MHPs 光催化析氢性能总结在表1 中。 目前取得的实验成果充分证明了MHPs 在光催化析氢方面的应用潜力,但纯组分的MHPs光催化效率及稳定性较低,通过构建异质结构、引入电荷传输通道、优化能级结构和表面配体密度、引入掺杂离子或采用全无机卤化物钙钛矿材料等策略可以进一步提升光催化析氢速率和稳定性。在提高光催化效率方面,提高光生载流子的分离和迁移效率是关键,需要限制光生载流子在迁移过程中的非辐射复合,缩短其到反应位点的迁移距离。 在提高稳定性方面,需要平衡催化速率与稳定性的关系,可以采用一些电导性良好的材料对MHPs 进行表面钝化,在提升其湿稳定性的同时不降低光生载流子向外传输的能力,例如TiO2、聚乙烯二氧噻吩等。 总的来说,MHPs 光催化析氢的应用研究尚处于初步阶段,其光催化速率和稳定性仍有待进一步提升。
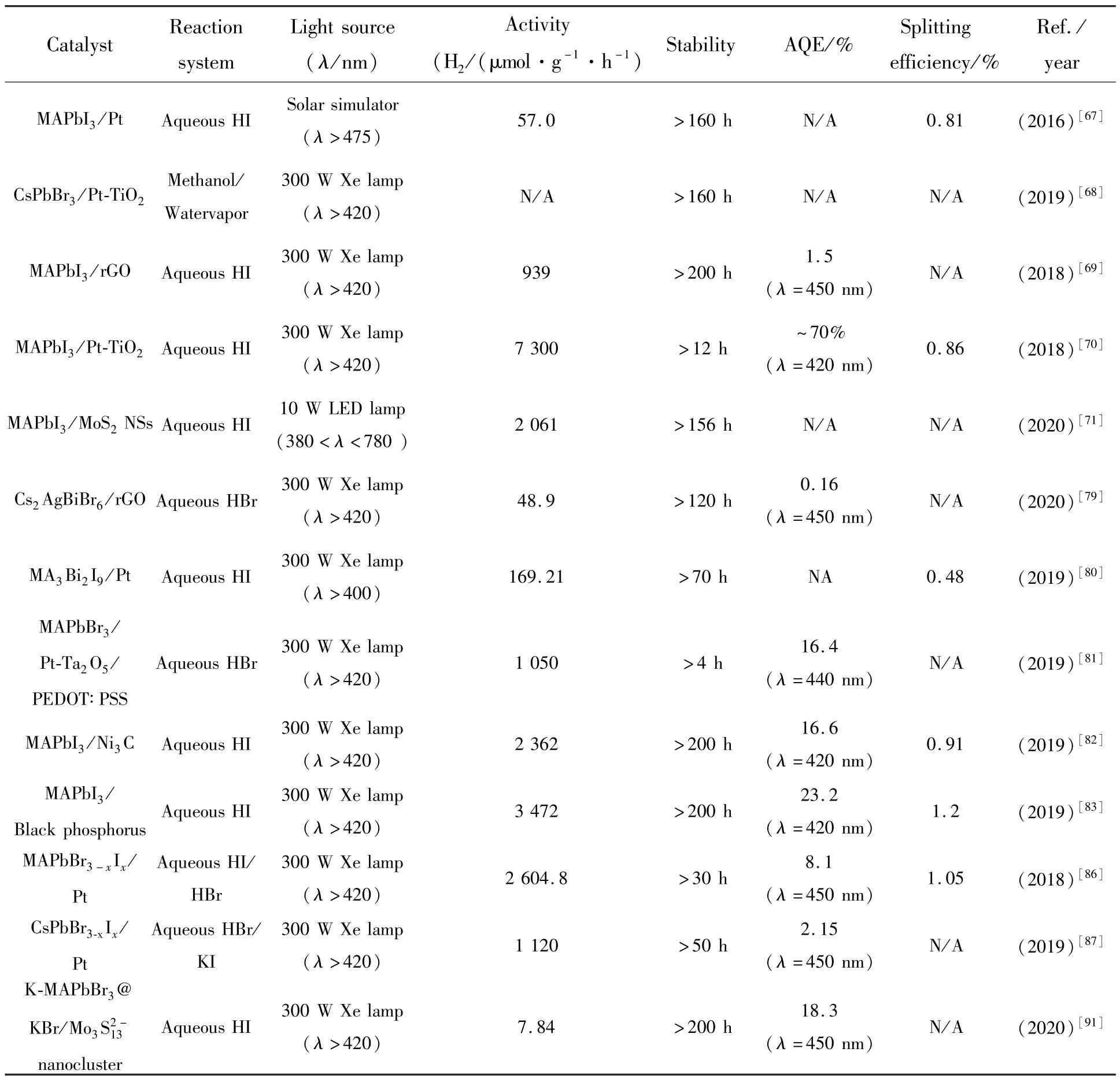
表1 MHPs 在不同实验条件下的光催化析氢性能Tab.1 Summary of the photocatalytic H2 evolution performances using metal halide perovskite under different experimental conditions
4.2 光催化CO2 还原
4.2.1 光催化CO2还原机理
光催化CO2还原的反应机理与光催化析氢类似,但线性排列的CO2分子具有较高的热力学稳定性(= -394.36 kJ/mol)[92],因此需要有足够的能量对其活化。 当CO2在光催化材料表面被活化形成活性物质后,通常存在两种不同的反应方式[93]:(1)活性物质先转化成CO 进而被还原为碳自由基C·,随后C·逐一与单电子和单质子作用,最后生成CH4;(2) 活性物质直接与多电子和多质子相互作用生成CO、HCOOH、HCOH、CH3OH、CH4等产物,作用式如(6) ~(10)所示:
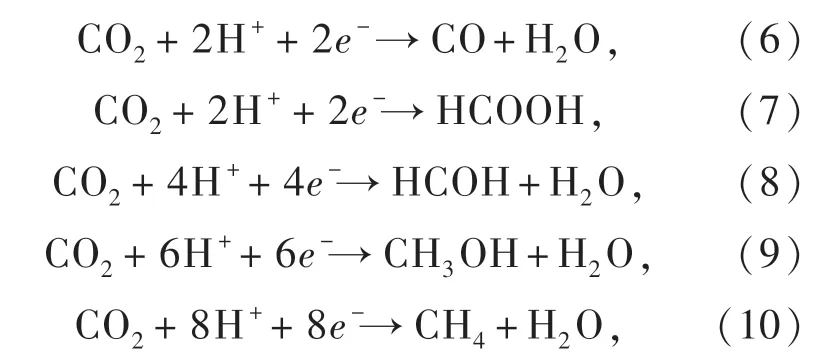
其相应的还原电势如图9(a)所示。 在CO2还原反应中,使一个CO2分子接受一个电子形成CO2/自由基需要克服较大的能量势垒( -1.9 Vvs. NHE,pH=7),而多电子和多质子的CO2还原反应则具有相对低的能量势垒,因而在热力学上更有利于反应的进行[94]。
通常,光催化CO2还原反应存在8 个动态过程,如图9(b)所示:(1)半导体的光吸收和光激发;(2)光生电子-空穴对的形成和向半导体表面迁移;(3)和(4)光生电子-空穴对的复合;(5)光生电子催化H2还原反应;(6)光生空穴催化CO2还原反应;(7)光生空穴催化H2O 发生氧化反应;(8)水氧催化还原产物进一步氧化。 为保障光催化CO2还原获得较高的速率和产率,需要抑制(3) ~(5)和(8) 这4 个反应过程,从而提高光生载流子的利用率。
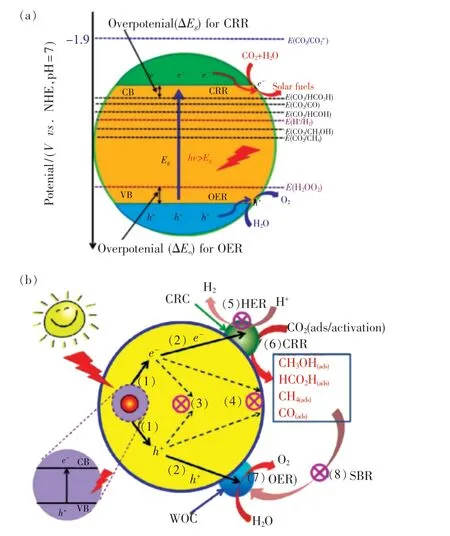
图9 (a)光催化CO2 还原机理;(b)光催化CO2 还原的过程[95]。Fig.9 (a)Schematic illustration of CO2 photoreduction on a semiconductor. (b) Process of CO2 photoreduction with water[95].
4.2.2 光催化CO2还原的研究进展
2017 年,Xu 等[74]率先报道了CsPbBr3QDs的光催化CO2还原的性能。 以乙酸乙酯/水作为溶剂,CsPbBr3QDs 光催化CO2还原速率为23.7 μmol·g-1·h-1,还原产物为CO、CH4、H2,其中CO2的选择性催化率超过99.3%。 通过将CsPb-Br3QDs 与石墨烯(GO)复合,促进了光生载流子的分离和传输,CsPbBr3QDs/GO 的电荷消耗速率相较于CsPbBr3QDs 提升了25.5%,如图10(a)、(b)所示。 几乎在同一时间,Hou 等[96]研究了不同粒径(3.8,6.1,8.5,11.6 nm)的CsPbBr3QDs对光催化CO2的影响。 实验表明,不同粒径的CsPbBr3QDs 的带隙和比表面积会对CO2的还原速率造成影响。 通过优化CsPbBr3QDs 的带隙和比表面积,粒径尺寸为8.5 nm 的CsPbBr3QDs 对CO2的选择性还原超过99%,平均电子消耗速率为20.9 μmol·g-1·h-1。 通过第一性原理计算发现,CsPbBr3QDs 相较于其块体材料,CsPbBr3QDs 的导带底和价带顶的电荷态密度更大,因此在太阳光辐照下,光生电子可以有效地被激发到导带底,从而提升了光化学转换效率,如图10 (c)、(d)所示。
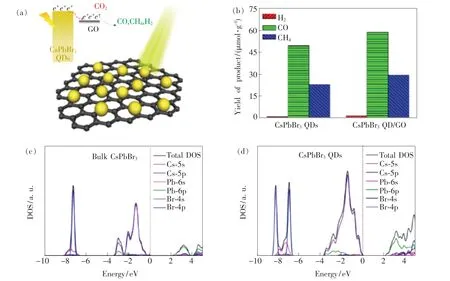
图10 (a)CsPbBr3 QDs/GO 光催化还原CO2 机理; (b)CsPbBr3 QDs 和CsPbBr3/GO 在12 h 内光催化CO2 还原的产物生成速率[74];块体CsPbBr3(c)和CsPbBr3 QDs(d)的态密度[96]。Fig.10 (a)Schematic diagram of CO2 photoreduction over the CsPbBr3 QDs/GO photocatalyst. (b)Photocatalytic performance:yield of the CO2 reduction products after 12 h of photochemical reaction[74]. Calculated densities of state diagrams of bulk CsPbBr3(c) and CsPbBr3 QDs(d)[96].
由于纯组分MHPs 对CO2的捕获能力较弱,导致还原速率较低。 通过界面工程构造异质结构,可以进一步促进内部光生载流子向外传输效率,增大CO2吸附面积,增强光催化材料的活化能力,提升MHPs 在湿度环境的稳定性等,因此常被用于提高MHPs 的光催化CO2还原速率。Kuang 等将CsPbBr3NCs 分别与二维Pd NSs 和无定形的TiO2组成异质结构,如图11(a)、(b)所示。 具有肖特基结的CsPbBr3/Pd NSs[97]与无定形TiO2包裹的CsPbBr3/a-TiO2[75]在光催化CO2还原反应中的电子消耗率均获得较大提升,分别比纯CsPbBr3NCs 提高了2.43 倍和6.5 倍,如图11(d)、(e)所示。 将介孔材料和二维材料与MHPs 组合成异质结构也是提升光催化CO2还原速率的有效方法。 Xu 等[72]将CsPbBr3QDs 锚定在富-NHx介孔g-C3N4NSs(PCN)上,如图11(c)所示。 一方面,CsPbBr3QDs 与PCN 两者间具有合适的能级排列,促进了光生载流子的分离和传输,如图11(f)所示;另一方面,PCN 与CsPbBr3QDs 在界面间形成N—Br 键,对CsPbBr3QDs 起到钝化作用,提升了光催化材料的稳定性。 优化CsPbBr3QDs 的用量,CsPbBr3QDs/PCN 的光催化CO2还原速率达到149 μmol·g-1·h-1,分别是g-C3N4纳米材料和CsPbBr3QDs 的3 倍和15 倍。类似地,Liu 等[98]通过原位生长法在Ti3C2TxNSs(MXene)的表面锚定CsPbBr3NCs,CsPbBr3/MX-ene 纳米结构显著提升了光生载流子的迁移效率。 合适的MXene 用量可使CsPbBr3/MXene 的电子消耗率达到110.6 μmol·g-1·h-1。 最近,Li 等[99]通过将过渡金属化合物2,2′∶6′,2″-三联吡啶(Ni(tpy))锚定在CsPbBr3QDs 的表面,组成CsPbBr3-Ni(tpy)复合结构。 由于Ni(tpy)中的聚氮苯基环可以捕获及储存电荷,且能够作为特定的催化活性位点,从而促进了CsPbBr3QDs 中的光生载流子迁移和光催化效率。 在可见光照射下,CsPbBr3-Ni(tpy)可以高效地将CO2还原为CO 和CH4,催化效率高达1 724 μmol·g-1,是纯CsPbBr3QDs 的26 倍。
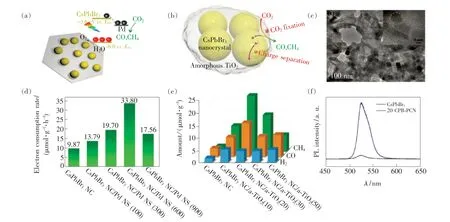
图11 (a)CsPbBr3/Pd NSs 光催化CO2 还原机理[97];(b)CsPbBr3/a-TiO2 光催化CO2 还原机理[75];(c)20% CsPbBr3 QDs/PCN 的形貌[72];(d)CsPbBr3 与CsPbBr3/Pd NS 光催化CO2 还原的电子消耗率对比[97];(e)CsPbBr3 与CsPb-Br3/a-TiO2 的光催化CO2 还原的产率对比[75];(f)CsPbBr3 QDs 与20% CsPbBr3 QDs/PCN 的荧光特性[72]。Fig.11 (a)Sketch of the composite material and their corresponding band alignments[97]. (b)A schematic illustration on the enhanced charge separation and CO2 fixation in a-TiO2-encapsulated CsPbBr3 nanocrystal[75]. (c)TEM image of 20 CPB-PCN[72]. (d)Electron consumption rates under visible light illumination( >420 nm)[97]. (E)Photocatalytic CO2 reduction test results[75]. (f)PL spectra of CsPbBr3 and 20 CPB-PCN[72].
通过调控卤素成分,可以优化光催化材料的能级结构与光催化氧化-还原电势相匹配,或与金属有机框架(Metal-organic frameworks,MOFs)组合,如ZIF-67[73]、UiO-66(NH2)[100]、PCN-221(Fex)[101]等,可以增加光催化材料与CO2的接触面积,同时起到一定的钝化作用,实现更好的稳定性和更高的催化效率。 Su 等[102]的研究表明,改变卤素成分影响着CO 和CH4的产率,通过调控合适的卤素配比(Br/Cl),CsPb(Br0.5Cl0.5)3NCs 的光催化CO2还原的效率相比于CsPbBr3NCs 和CsPbCl3NCs分别提高了4.5 倍和9.1 倍。 目前光催化CO2的反应体系常将MHPs 放入某些特殊溶剂中以提升其湿稳定性,相比于用水作为溶剂,这样的光催化体系通常会造成有价值的溶剂不必要的浪费,不利于实际的应用。 最近,Zhang 等[103]采用甲基丙烯酸六氟丁酯(HFBMA)作为疏水层,通过Co2+掺杂制备了Co@CsPbBr3/Cs4PbBr6,由于HFBMA在水中会逐渐发生水解形成双亲性六氟丁醇,使得Co@CsPbBr3/Cs4PbBr6具有较好的水溶性,如图12(a)所示。 通过对比掺杂前后的能级结构发现(图12(b)),Co2+能够从CsPbBr3/Cs4PbBr6获取光生电子,进而将CO2还原成CO 和CH4。 掺杂浓度为2%的Co@CsPbBr3/Cs4PbBr6在纯水条件下将CO2还原为CO 和CH4的平均催化速率为12.35 μmol·g-1·h-1,且可连续光催化超过20 h,如图12(c)所示。 随后,他们还探究了其他金属离子(Co2+,Ni2+,Fe3+)掺杂CsPbBr3/Cs4PbBr6对光催化CO2还原的影响,如图12(d)所示。 采用金属离子掺杂和疏水层保护的策略很好地解决了MHPs 湿稳定性问题,但疏水层同时也降低了CO2与还原位点的接触,使得光催化速率较低,因此仍需对该反应体系进行优化。
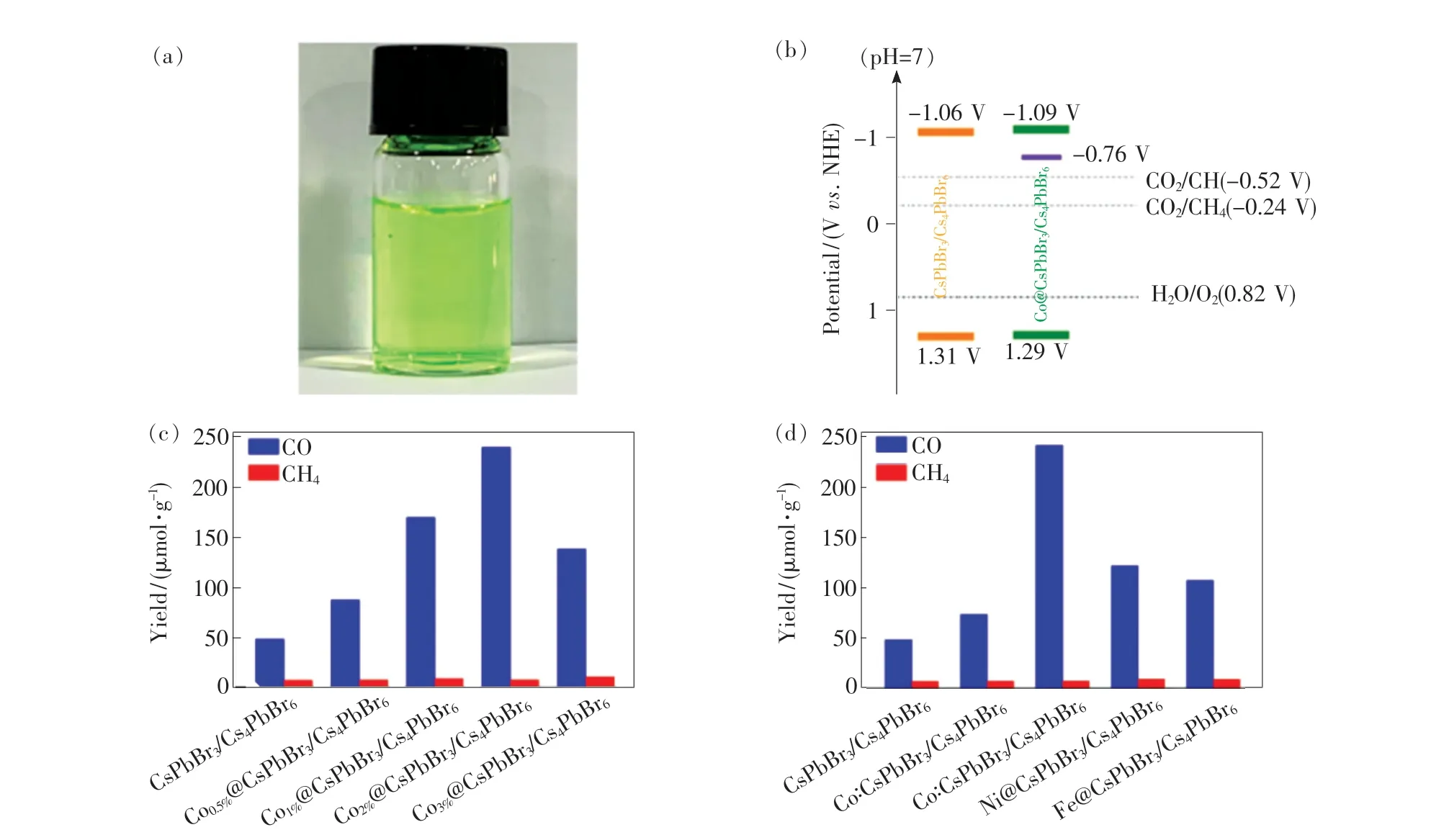
图12 (a)Co@CsPbBr3/Cs4PbBr6 在纯水中的分散情况;(b)CsPbBr3/Cs4PbBr6 和Co@CsPbBr3/Cs4PbBr6 NCs 的能带结构;(c)CsPbBr3/Cs4PbBr6 和Co@CsPbBr3/Cs4PbBr6 NCs 光催化CO2 还原的产率比较;(d)掺杂不同阳离子的产率比较[103]。Fig.12 (a)Optical image of Co@CsPbBr3/Cs4PbBr6 in pure water. (b)Band structures for CsPbBr3/Cs4PbBr6 and Co@CsPb-Br3/Cs4PbBr6 NCs derived from the LSV and UV/Vis diffuse reflectance spectroscopy measurements. The yields of CO and CH4 generated from photocatalytic CO2 reduction in pure water based on undoped and Co-doped CsPbBr3/Cs4PbBr6 with various doping concentrations(c) and pristine CsPbBr3/Cs4PbBr6, Co∶CsPbBr3/Cs4PbBr6 and CsPbBr3/Cs4PbBr6 with mixing different metal cations(d) as photocatalysts after 20 h of irradiation under 300 W Xe-lamp, with the light intensity of 100 mW· cm -2[103].
在无铅卤化物钙钛矿方面,Kuang 等对Cs2AgBiBr6NCs[104]和Cs2SnI6NCs[105]的光催化CO2还原性能进行了研究。 通过热注射法制备的立方相Cs2AgBiBr6NCs 具有良好的光、热、湿稳定性,将其应用于光催化CO2反应还原,在可见光照射6 h 内可以光催化CO2还原为CO 和CH4,催化产率分别为14.4 μmol·g-1和9.6 μmol·g-1。实验发现,通过洗脱步骤降低Cs2AgBiBr6NCs 表面的配体密度,可以提高内部光生载流子向外传输的能力,减少产物和中间产物在表面配体层积累,使得Cs2AgBiBr6NCs 的总电子消耗率提高了6.5 倍,达到105 μmol·g-1。 通过简单的液相法原位合成了Sn 原子共享的Cs2SnI6/SnS2Ⅱ型异质结,延长了光生电子在SnS2的寿命,促进了光生载流子的分离和迁移效率,使得Cs2SnI6/SnS2在3 h 内光催化CO2还原的效率(6.09 μmol·g-1)较纯组分Cs2SnI6提升了5.4 倍。 通过共享原子原位构造异质结构的策略对于其他类型的钙钛矿结构具有普适性,但由于Cs2SnI6本身的载流子迁移速率较低,因此后期仍需要注重发展有效的策略提升其光生载流子的分离和迁移效率。Geyer 等[106]采用热注射法制备了尺寸均匀的Cs3Sb2Br9NCs,在制备过程中采用辛酸替换油酸是获得高质量Cs3Sb2Br9NCs 的关键。 Cs3Sb2Br9NCs 光催化CO2还原反应可以得到单一产物CO,在4 h 内的光催化效率相比于CsPbBr3NCs 提升了10 倍,达到510 μmol·g-1·h-1。
表2 总结了一些目前MHPs 光催化还原CO2的性能。 综上所述,MHPs 光催化CO2还原的应用近年来取得了不错的研究成果。 由于自然状态下CO2为气体状态,为增加CO2与光催化材料的接触面积,在光催化反应体系中常将CO2溶于溶剂中,但由于MHPs 的湿稳定性较差,通常采用无极性或弱极性溶剂作为CO2的溶解剂,如乙酸乙酯、乙腈、异丙醇等,且选择湿稳定性较好的全无机金属卤化物钙钛矿材料作为光催化材料。 目前MHPs 光催化CO2的还原产物多为混合状态,因此需要提高反应底物的光催化选择性以及单一还原产物产率,以降低后期产物分离带来的成本支出,提高经济效益。 通过不断优化催化结构,促进光生载流子向催化位点迁移,增加光催化材料中的还原位点,提高光催化体系对CO2的吸附等,可以进一步提升光催化材料还原效率。 在提升MHPs 湿稳定性的同时需要保证其对CO2的吸附能力,可以采用疏水层钝化与助催化剂结合的策略,例如沉积疏水层后进行挖孔填充助催化剂,可以极大地平衡MHPs 的抗水性和光催化效率。 总的来说,MHPs 光催化CO2还原在催化效率、单一目标产物的选择性及光催化稳定性等方面仍有较大的提升空间。
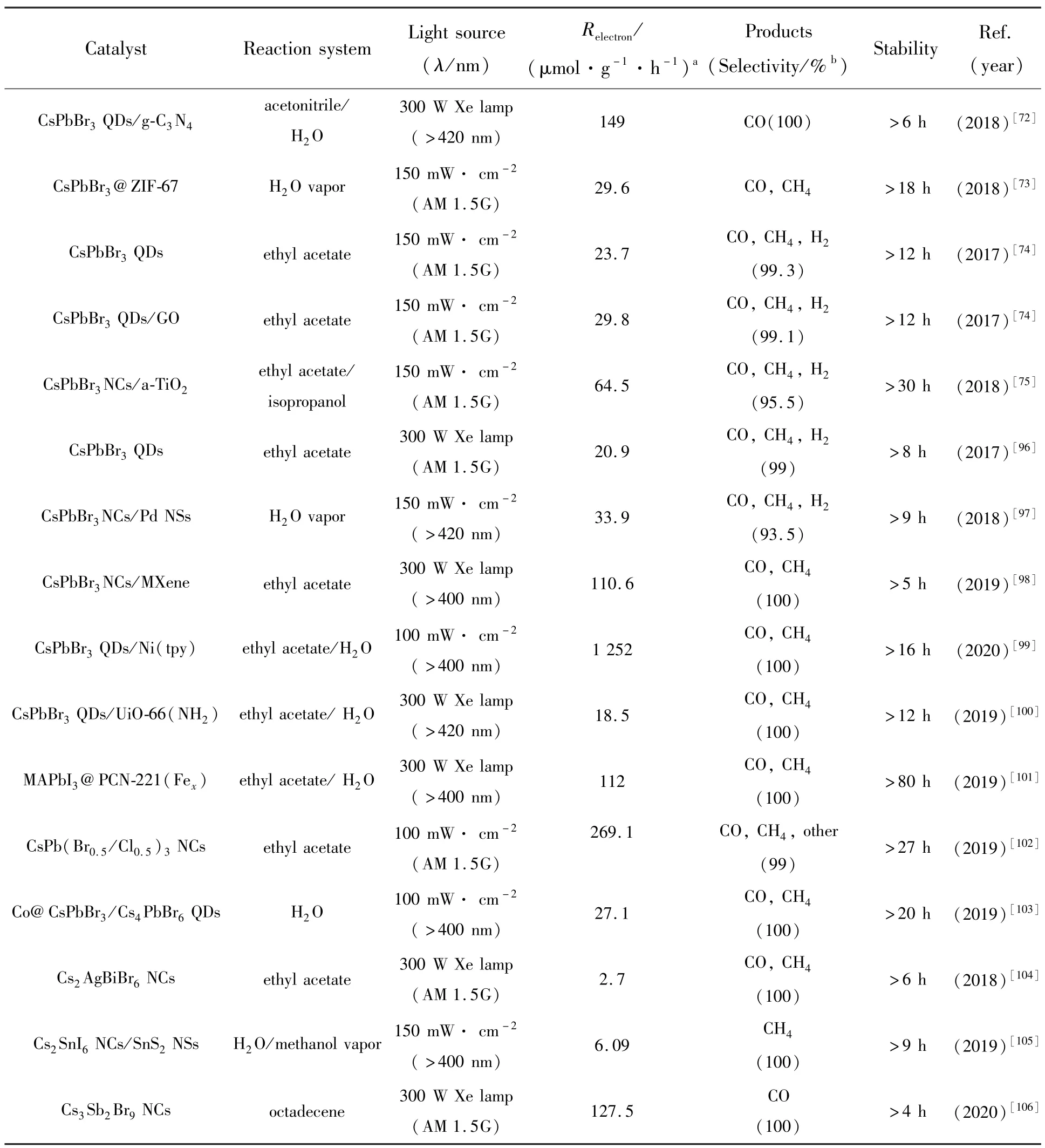
表2 MHPs 在不同实验条件下的光催化CO2 还原性能Tab.2 Summary of the photocatalytic CO2 reduction performances using metal halide perovskite under different experimental conditions
4.3 光催化有机物转化
MHPs 在有机物转化中主要分为两个方向:一是光催化有机物化学键形成,得到高附加值的目标产物;二是光催化有机污染物化学键断裂,使其降解为CO2、H2O、小分子化合物或无机盐等,降低其对生态环境的危害。
4.3.1 光催化有机物合成
有机物的聚合反应是合成高附加值聚合物的常用手段。 2017 年,Chen 等[107]首次报道了CsPbI3QDs 光催化三聚体3,4-乙烯二氧噻吩(TerEDOT)的聚合反应,采用1,4-苯醌或氧分子作为电子受体,在可见光照射下,光生空穴迁移到噻吩衍生物,使TerEDOT 发生氧化并进行聚合反应合成聚3,4-乙烯二氧噻吩(PEDOT),如图13(a)所示。通过能级分析可知(见图13(b)),由于3,4-乙烯二氧噻吩(EDOT)的氧化电势位于CsPbI3QDs 价带顶的下方,因此CsPbI3QDs 不能对单体EDOT直接进行光催化聚合,通过增加EDOT 主链长度可以使得TerEDOT 的氧化电势位于带隙中间。此外,由于聚合反应生成的PEDOT 具有一定的导电性,在PEDOT 对CsPbI3QDs 表面进行包覆提升其稳定性的同时也可以平衡光电器件应用领域中的电荷传输问题,如图13(c)所示。 基于光催化聚合反应对MHPs 进行原位封装提升稳定性的策略,Tan 等[108]通过CsPbBr3NCs 在白光LED 照射下光催化聚合苯乙烯,生成的聚苯乙烯分子质量可以高达200 ku,如图13(d)所示。 表面高聚合物的包覆提高了CsPbBr3NCs 的光致发光量子产率和湿稳定性,使其薄膜和LED 器件获得了很好的抗水性。 此外,CsPbBr3NCs 光催化聚合甲基丙烯酸甲酯(MMA)的可行性也得到了验证,连续反应12 h 后的PMMA 的转化率达到25%。 通过光催化有机物聚合的方式对MHPs 进行包裹,提高其对环境因素的容忍性,有望进一步提高其在光电器件领域遇到的性能瓶颈问题。
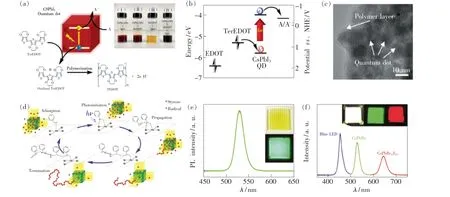
图13 (a)CsPbI3 QD 光催化聚合TerEDOT 机理;(b)EDOT、TerEDOT 和CsPbI3 QDs 的能级结构;(c)PEDOT 包裹CsPbI3 QDs 后的形貌[107];(d)CsPbBr3 NCs 光催化苯乙烯聚合反应机理;(e)CsPbBr3 NCs-聚苯乙烯的发光特性;(f)基于CsPbBr3 NCs-聚苯乙烯构成的LED 发光光谱[108].Fig.13 (a)Illustration of the proposed mechanism for photocatalytic polymerization of TerEDOT over CsPbI3 QD under visible light illumination. A indicates the electron acceptor. (b)Energy diagram of EDOT,TerEDOT and CsPbI3 QDs. The arrows indicate the electron transfer direction. (c)TEM image of CsPbI3 QDs encapsulated by PEDOT[107]. (d)Proposed reaction mechanism of the perovskite photoactivated polymerization of styrene. (e)PL spectrum and inset images of a perovskite-polystyrene nanocomposite film prepared after a 14 h photopolymerization reaction. (f)Spectral characteristics and inset image of red and green-emitting perovskite-polystyrene nanocomposite films and their combination with blue LED to form a white light-emitting downconversion device[108].
除了光催化有机物聚合反应,MHPs 可以光催化合成多种化学键。 Wu 等[109]采用CsPbX3QDs 实现了光催化硫醇有机物的偶联反应,高效的S—H 键活化使二硫化物获得了较高的产率(68% ~96%),且通过对C—H 键活化,实现了叔胺与亚磷酸酯的脱氢C—P 偶联反应。 Yuan等[110]构建FAPbBr3/TiO2复合结构,实现了对苄基醇的高效选择性光催化氧化,其作用机理如图14(a)所示。 在可见光照射下,FAPbBr3产生的光生电子向TiO2转移,并进一步与TiO2表面的O2结合形成·O2- 自由基,而光生空穴与苄基醇(R—CH2OH)结合形成碳正离子(R—CH2OH*+),最后碳正离子与·O2-自由基相互作用合成醛类物质并释放H2O 分子。 实验表明,TiO2的引入促进了光生载流子的选择性迁移,使光催化苄基醇的醛类氧化具有较高的效率和选择性,FAPbBr3/TiO2的光催化效率是纯FAPbBr3的4 倍。 类似的活性增强现象同样发生在CsPbBr3/TiO2对苄基醇的光催化氧化反应中[111]。 最近,Zhu 等[112]报道了CsPbX3NCs 和MAPbX3NCs 光催化C—C键的直接形成。 通过调控反应条件,CsPbX3NCs和MAPbX3NCs 可以在有机溶剂中选择性地催化多种重要反应,如醛类的α-烷基化反应、sp3-C 偶联反应和卤代烷烃脱卤还原反应等。 随后,他们实现了C—C 键、C—N 键和C—O 键的MHPs 光催化合成,且通过调MHPs 的带隙可以进一步扩大光催化底物的范围[113]。 最近,Tüysüz 等[114]报道了无铅钙钛矿Cs3Bi2Br9与介孔SBA-15 SiO2复合的光催化性能。 实验表明,Cs3Bi2Br9/SBA-15 在可见光照射下对脂肪族和芳香族烃类有机物的C(sp3)—H 键具有高效的催化活性,其中转化效率高达32 900 μmol·g-1·h-1,该转化效率远超过目前已有的光催化材料,且催化选择性超过99%。
4.3.2 光催化有机物降解
2017 年,Xu 等[115]报道了CsPbX3QDs 对甲基橙(Methyl orange,MO)的光催化降解性能。 实验表明,CsPbCl3和CsPbBr3QDs 都对MO 的降解表现出较高的催化活性。 如图14(b)所示,随着反应的进行,溶液中MO 的颜色逐渐变浅,2 mg CsPbCl3在80 min 内可以降解90%的甲基橙(3 mL,10 mg·mL-1)。 此外,在相同条件下,1 mg CsPbBr3在100 min 内的降解效率为89%。 最近,Fan 等[116]利用CsPbX3QDs 在乙醇中降解抗生素盐酸四环素(Tetracycline hydrochloride,TC-HCl)和MO。 结果表明,在100 mL 的乙醇溶液中,100 mg CsPbBr3在30 min 内对TC-HCl 和MO(100 mL,10 mg·L-1) 的降解效率分别为76% 和70%。 通过X 射线衍射实验发现,CsPbBr3在乙醇中具有较高的稳定性,如图14(c)所示。 Liu等[117]通过组分工程制备了CsPb(Br1-xClx)3NCs,并将其与AuNCs 复合构成纳米异质结构,如图14(d)所示。 光激发下,在CsPb(Br1-xClx)3和Au 之间形成内建电场,促进了光生载流子的分离和迁移,通过溶解的氧气对光生电子的捕获生成超氧自由基·O2-,同时Au NCs 增强了羟基自由基·OH 的形成,使得CsPb(Br1-xClx)3-Au 在6 h内降解了71%的苏丹红Ⅲ(20 mL,2. 43 mol·mL-1),降解速率比纯组分CsPb(Br1-xClx)3提高了3 倍以上。 Xu 等[118]采用湿化学法合成MASnI3/TiO2异质结构,将其用于降解染料罗丹明B。 实验表明,MASnI3/TiO2在40 min 内可降解97%的罗丹明B。 但由于Sn2+极易被氧化为Sn4+,需要提升其抗氧化性以提高锡基钙钛矿光催化材料的可重复利用率。 最近,Hu 等[119]创新性地将纳米银、CsPbBr3QDs、块体g-C3N4(CN)组合成三元体系结构Ag-CsPbBr3/CN,如图14(e)所示。 在可见光条件下,Ag-CsPbBr3/CN 的光催化降解性能相较于其他组分得到了极大的提升,纳米银质量分数为7%的组分可以在140 min 内有效降解7-氨基头孢菌素酸溶液,降解速率常数kobs高达18.97 ×10-3min-1,如图14(f)所示。 此外,7%Ag-CsPbBr3/CN 同样可以高效降解头孢克肟和头孢曲松钠,140 min 内的降解效率分别为94.23%和93.41%。 光催化降解效率的提高来源于增强了光催化材料对反应底物的吸附能力及三元体系结构对光生载流子分离和迁移的促进作用。
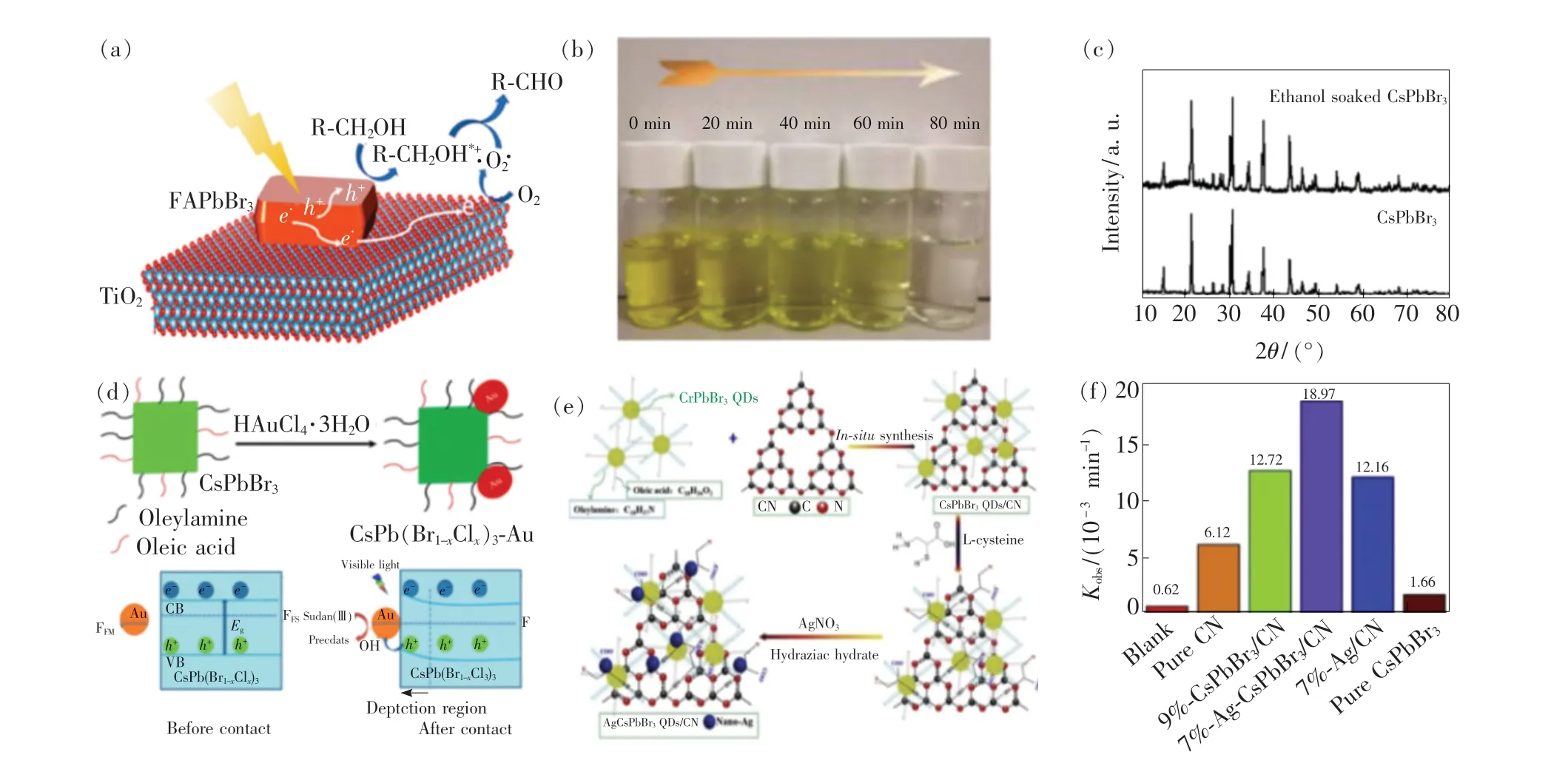
图14 (a)FAPbBr3/TiO2 光催化氧化苄基醇的机理[110];(b)CsPbCl3 QDs 降解MO 的过程[115];(c)CsPbBr3 加入乙醇前后XRD 对比[116];(d)CsPb(Br1-xClx)3-Au 的制备示意图及其光催化机理[117];(e)三元体系Ag-CsPbBr3/CN 的合成过程;(f)不同组分的光催化降解速率[119]。Fig.14 (a)Corresponding schematic of the proposed selective photocatalytic benzyl alcohol to benzaldehyde oxidation process over the FAPbBr3/TiO2 hybrid[110]. (b)Image of MO degradation using CsPbCl3 QDs[115]. (c)XRD pattern of fresh and ethanol soaked CsPbBr3[116]. (d)Preparation diagram of CsPb(Br1-xClx)3-Au nanoheterostructures and the mechanisms for the photocatalytic degradation of Sudan Red Ⅲ[117]. (e)Synthetic process of the Ag-CsPbBr3/CN ternary assembly(not to scale). (f)Photocatalytic degradation rate constant k[119].
综上所述,MHPs 在光催化有机物的合成及降解的应用中表现出优越的性能,但目前仍面临着诸多问题与挑战,如催化效率、催化稳定性和催化产物提纯等。 在光催化效率方面,提高光生载流子的利用率是关键。 可以通过引入助催化剂或构造异质结界面,一方面,可以促进光生电子-空穴对的分离和迁移;另一方面,可以增强光催化材料对反应底物的吸附能力及光催化材料的稳定性。 在催化稳定性方面,光催化有机物转化的反应条件常涉及水相和极性有机溶剂,仅通过表面配体包覆的方式不足以提升MHPs 的长期稳定性,需要进一步提出解决稳定性的优化方案。 在催化产物提纯方面,目前对这方面的研究报道较少,可以考虑通过化学分离提纯的方式,对溶液中的产物逐一分离。 整体来说,MHPs 在光催化有机物转化方面的研究仍需进一步深入,其催化效率、催化稳定性、催化底物的广延性等仍有较大的提升空间。
5 总结与展望
MHPs 优异的光电特性使其近年来在光催化析氢、光催化还原CO2及光催化有机物转化等方面都取得了不错的研究进展。 目前,不同课题组所采取的光催化实验手段和评判方法不尽相同,对于评判光催化效果带来一定的差异。 总的来说,MHPs 的光催化应用研究仍处于初步阶段,其光催化效率、稳定性、目标产物选择性及催化底物的广延性等光催化性能仍有很大的提升空间。 提高MHPs 的光催化性能,其核心在于提高光生载流子的利用率和MHPs 的环境稳定性。 针对光生载流子的利用率问题,一方面可以采用助催化剂掺杂或构造表面异质结构等策略,促进光生载流子的分离和迁移效率,增加光催化材料与反应底物的接触面积和吸附能力;另一方面可以通过原位钝化和等效配体等策略减少制备过程中引入的缺陷态,抑制光生载流子在迁移过程中的缺陷态复合。 针对MHPs 环境稳定性问题,可以通过组分工程优化钙钛矿的晶体结构,或采用无极性或低极性的溶剂作为反应溶剂,降低环境因素(光、氧、水分等)对结构造成的破坏。 此外,可以从平衡光生载流子利用率和环境稳定性的角度考虑,选择合适的材料对MHPs 进行钝化处理,以提升环境稳定性。 针对反应底物的广延性问题,可以考虑卤素替换或量子限域效应策略,扩宽MHPs的带隙宽度,使得更多反应底物的氧化-还原电势处于带隙中成为可能。 同时,考虑到光催化聚合某些特性有机物时可以采用光催化的方式对低维钙钛矿材料进行包覆处理,如纳米片和纳米线,以解决其在光电器件应用中稳定性差的困扰,从而扩宽MHPs 光催化的应用范围。 相信随着MHPs在光电领域的不断深入发展,MHPs 在化工能源、环境治理、绿色有机化学和光电器件等领域都将具有广阔的应用前景。 通过合理的调控手段,MHPs 有望成为新一代高效率、高经济性、高普适性的光催化材料。
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
物理学报(2022年6期)2022-03-30
陶瓷学报(2021年2期)2021-07-21
石油化工高等学校学报(2021年3期)2021-07-15
物理学报(2020年16期)2020-08-29
科技风(2018年9期)2018-05-14
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
原子能科学技术(2015年8期)2015-12-15
太阳能(2015年4期)2015-02-28
