
黄芪多糖对C3H10T1/2细胞棕色脂肪分化中长链非编码RNA表达谱的影响
2020-09-11 07:40张世和曹宇鑫邓步皓高旭阳赵俊星
激光生物学报 2020年4期
张世和,曹宇鑫,邓步皓,高旭阳,赵俊星
(山西农业大学动物科技学院,太谷 030800)
黄芪多糖(astragalus polysacharin,APS)提取自中药材黄芪的根部,作为黄芪最重要的天然有效成分之一发挥作用。随着人们对多糖研究的加深,其在抗氧化、抗炎、免疫调节、抗肿瘤、降血脂、降血糖等多方面的生物活性与功能也随之被发现[1-4]。APS也因其能促进3T3-L1前脂肪细胞的增殖和分化[5]以及抑制营养消化和吸收酶的活性来治疗肥胖症而备受关注[6]。近年来的研究多将APS运用于2型糖尿病、肥胖和代谢综合征等代谢相关性疾病的防治上。
棕色脂肪(brown adipose tissue,BAT)是哺乳动物发热生热的关键部位,其产生的热量对于初生哺乳动物在寒冷环境中的生存及冬眠动物的体温维持至关重要。BAT由于含有大量的线粒体以及解耦联蛋白 1(uncoupling protein 1,UCP1)成为非寒颤性热发生的主要组织,从而增加能量消耗[7]。近年来的研究表明,BAT能通过摄入甘油三酸酯来加速血浆脂质的清除,从而改善血脂异常和胰岛素抵抗[8]。在人和小鼠中的研究均发现,BAT可积极利用线粒体中的支链氨基酸进行热发生,进而控制肥胖的发生[9-10]。因此,通过激活BAT来增加能量消耗已成为一种安全的预防和控制肥胖的方法。
长链非编码 RNAs(long non-coding RNAs,lncRNAs)参与不同的生物学过程[11],并在器官发育过程中发挥着关键作用,包括细胞增殖、细胞分化、细胞周期的调节、凋亡和衰老[12]。近年来的研究发现,lncRNAs具有调节脂肪形成的能力[13-14],并鉴定出多个与棕色脂肪发育相关的lncRNAs。lncRNA-Blnc能与早期B细胞因子2(recombinant early B-cell factor 2,EBF2)形成核糖核蛋白复合体来增强EBF2的表达,并通过调节产热基因来促进小鼠米色和棕色脂肪细胞的分化[15]。lncBATE1能维持棕色脂肪细胞核心标记基因的表达,抑制白色脂肪标记基因,且能与核异质核糖核蛋白U(heterogeneous nuclear ribonucleoprotein U,hnRNP U)相互作用以促进米色和棕色脂肪的形成[16]。
本研究以小鼠间充质干细胞C3H10T1/2为研究对象,在体外培养的过程中添加APS,收集样品并进行转录组测序分析,旨在揭示APS促进棕色脂肪细胞C3H10T1/2分化的作用机理,并阐明lncRNAs在棕色脂肪细胞分化中的功能。
1 材料与方法
1.1 细胞和主要试剂
C3H10T1/2细胞购买自美国模式培养物集存库(American type culture collection,ATCC);达尔伯克改良伊格尔低糖培养液(Dulbecco’s modified eagle medium,DMEM,SH30021.01)购自于 Hyclone公司;双抗(青链霉素,10378016)与胎牛血清(10091155)购自于Gibco公司;三碘甲腺原氨酸(3′-triiodothyronine,T3)、吲哚美辛、胰岛素(I5500)、地塞米松(dexamethasone,Dex,D4902)和 3-异丁基 -1-甲基黄嘌呤(3-isobutyl-1-methyl-7H-xanthine,IBMX,I7018)购自于美国Sigma公司;PrimeScriptTMRT Master Mix(R047A)与 SYBR®Premix ExTaqTMII65(R820A)购于日本TaKaRa公司。
1.2 C3H10T1/2细胞的培养和棕色化诱导
C3H10T1/2细胞种植在生长培养基(含2%双抗、10%胎牛血清的低糖DMEM)中,在37℃、5%CO2的细胞培养箱中培养。待细胞铺满培养皿后,分别使用未添加和添加0.4 g/L APS的分化培养基(0.5 mmol/L IBMX、5 μmol/L Dex、1 nmol/L T3、0.125 mmol/L 吲哚美辛和10 g/L胰岛素溶于含有2%双抗的10%胎牛血清的DMEM培养基中)诱导分化,分化1.5 d后取样用于后续试验。
1.3 cDNA文库的构建和测序
以未经APS处理的棕色脂肪细胞为对照组,经0.4 g/L APS处理为试验组,每组做3个生物学重复,构建6个cDNA文库进行测序。上机前样品提取总RNA后,将rRNAs去除以保留mRNAs和ncRNAs。使用片段缓冲液将得到的mRNAs和ncRNAs片段化为短片段,并用随机引物将其逆转录为cDNA第一链。接着加入缓冲液、dNTPs(dUTP代替dTTP)、核糖核酸酶 H(ribonuclease H,RNase H)和 DNA polymerase I合成cDNA第二链。通过使用QiaQuick PCR试剂盒纯化cDNA片段进行末端修复,并加碱基A和Illumina测序接头。然后使用尿嘧啶-N-糖基化酶(Uracil-N-glycosylase,UNG)降解第二链 cDNA。通过琼脂糖凝胶电泳选择降解产物的大小,进行PCR扩增。最后通过Illumina HiSeqTM4000平台对文库制备物进行测序。
1.4 测序数据过滤
利用 FastQC(v0.11.4)(http://www.bioinformatics.babraham.ac.uk...jects/fastqc/)分析了reads的碱基组成、质量分布以及GC和AT的碱基含量。通过删除全为A碱基、具有10%以上N碱基和质量值(Q≤20)超过整条reads 50%的低质量reads,来得到高质量的reads用于后续的信息分析。
1.5 比对分析及重构转录本
使用reads比对工具bowtie2[17](2.2.8)和比对软件Tophat2[18](2.1.1)将高质量的reads比对到该物种的核糖体中(错配数:0),去掉比对上的reads后再比对到物种的参考基因组上。保留比对上的数据,使用Cufflinks[19]来重构转录本,根据组装出来的转录本在参考基因组上的位置以及筛选标准(转录本长度≥200 bp且exon数目≥2)筛选新的转录本,从而得到样本已知的和新的转录本。
1.6 新lncRNAs预测
对筛选出来的新转录本进行lncRNAs预测。使用CPC[20]和CNCI[21]2个软件预测其是否具有编码蛋白的能力,同时到蛋白数据库SwissProt中进行比对,取预测无编码能力且在蛋白数据库中无法比对上的转录本作为新lncRNA。
1.7 差异表达基因的分析
使用edgeR软件(http://www.r-project.org/)对组间lncRNAs和mRNAs的表达量进行差异分析,利用P值与log2FC来筛选差异mRNAs转录本,筛选条件为P<0.05且|log2FC|>1。
1.8 GO功能注释和KEGG通路分析
为了解lncRNAs靶基因的功能作用,我们使用基因本体(gene ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析对其进行了研究。GO总共有3个本体,分别描述转录本的分子功能、细胞组分和参与的生物过程。KEGG是分析基因在细胞中的代谢途径的主要公共数据库[22],尤其适用于分析基因组测序和其他高通量技术,从而得到大规模的数据。
1.9 通过Real-PCR验证基因和lncRNAs的表达
使用Trizol®Reagent试剂盒提取总RNA,提取后用1%琼脂糖凝胶电泳检测其是否降解,并用NanodropND-1000进行浓度与完整性检测,选取OD260nm/OD280nm为1.9~2.0者用于后续试验。RNA经DNA酶处理后,取1 μg进行cDNA反转录,并进行qRT-PCR分析。扩增程序为:95℃预变性20 s;95℃变性20 s,60℃退火及延伸20 s,36个循环。结果根据2-ΔΔCT法计算,18S rRNA基因为内参基因,引物序列见表1。
2 结果与分析
2.1 数据的来源
从Illumina HiSeqTM4000平台的每个文本库中平均产生了15 177 607 700个清洁数据,在去除不符合的碱基比例和低质量的reads后,从每个库中平均获得了14 831 667 631个高质量的reads。其中,测序准确率为99.9%的碱基数目占比可以达到95.19%~96.34%(表2)。在去除比对上的核糖体reads后,将其比对到小鼠的参考基因组上,有87.76%~90.65%的reads被成功定位到小鼠的参考基因组上(表2)。使用Cufflinks根据比对结果来重构转录本,共得到了 13 450个 lncRNAs和 57 776个 mRNAs。使用CPC、CNCI及蛋白数据库SwissProt等工具评估新转录本的蛋白质编码潜力,并取结果相交项作为最终结果,我们预测出了586个新lncRNAs转录本(图 1a)。

表1 引物序列Tab.1 Primer sequences for real-time qPCR
2.2 lncRNAs数据的分布和质量分析
根据lncRNAs在基因组上相对于蛋白编码基因的位置,我们将lncRNAs分为5种类型,其中有13 319个为基因间区型lncRNAs,16个为双向型lncRNAs,39个为反义 lncRNAs,63 个为正义 lncRNAs,13个为其他类型的 lncRNAs(图 1b),基因间区lncRNAs是最常见的lncRNAs类型,其比例也最大。在所有的lncRNAs中,大多数lncRNAs具有2个外显子(最多有27个外显子,平均3.10个外显子),显著低于蛋白质编码基因(最多有347个外显子,平均9.51个外显子)(图1c)。总体而言,lncRNAs的平均长度为1 251.84 bp,小于mRNAs的2 615.63 bp,lncRNAs和mRNAs长度的分布是一致的,相对较长的mRNAs转录物数量高于lncRNAs(图1d),转录本的平均表达值计算方法为每100万map上的reads中map到转录本上的每1 000个碱基上的fragment数目(fragments per kilobase of transcript per million mapped reads,FPKM),FPKM 小提琴图表明(图 1e),mRNAs的FPKM值为6.35,lncRNAs的FPKM值为1.75,mRNAs相对表达水平高于lncRNAs。
2.3 成对重复性检验
为获得对试验结果可靠性和操作稳定性的评估,我们对2次生物学平行试验的结果进行了相关性分析。lncRNAs的分析结果显示,样本间皮尔森相关系数(pearson correlation coefficient,PCC)全部大于0.990 0,最高可达到0.999 7。 mRNAs分析结果显示,样本间PCC全部大于0.990 0,最高为0.999 0,说明测序具有可重复性,试验结果的可靠性和操作的稳定性高(图2)。
2.4 组间差异表达基因分析
依据lncRNAs和mRNAs表达的火山图,我们发现了153个差异表达的lncRNAs(differentially expressed lncRNAs,DElncRNAs),其中有 76 个上调,77个下调(图3a、3c),同时也发现了1 238个差异表达的 mRNAs(differentially expressed mRNAs,DEGs),其中有590个上调,648个下调(图3b、3c)。随后我们对这些DEGs进行了GO功能注释和KEGG分析,GO功能注释结果显示(图3d),DEGs富集在了53个条目中,在3个本体的数量分布为:参与的生物过程24个、分子功能10个及细胞组分19个。参与的生物过程本体的条目主要有代谢过程、生长过程、细胞过程、生殖过程、生物过程调控等,其中细胞过程富集基因最多,有734个;分子功能本体的条目有结合、转录因子活性、蛋白质结合、核酸结合转录因子活性、分子功能调节剂、抗氧化活性等,其中结合功能富集基因最多,有645个;细胞组分本体的条目有细胞成分、突触、核苷酸、细胞外基质、细胞膜部分等,其中细胞成分富集基因最多,有753个。KEGG分析显示(图3e),这些基因富集在了343条通路中,根据富集显著性进行排序,排名前20的通路为:系统性红斑狼疮、酗酒、赖氨酸降解、癌症中的转录失调、皮质醇合成与分泌、破骨细胞分化、紧密连接、肿瘤坏死因子(tumor necrosis factor,TNF)信号通路、醛固酮合成与分泌、NOD样受体(nucleotide binding oligomerization domain-like receptors,NLR)信号通路、库欣综合征、甲状旁腺激素的合成分泌及作用、基底切除修复术、脂肪酸代谢、柠檬酸循环、局灶性粘连、人类嗜T细胞病毒I型(human T-cell leukemia virus-I,HTLV-I)感染、促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)信号通路、环磷酸鸟苷酸-蛋白激酶G(cyclic guanosine monophosphate-protein kinase G,cGMP-PKG)信号通路和代谢途径。其中TNF信号通路、脂肪酸代谢和柠檬酸循环等通路能积极参与BAT细胞分化的过程。
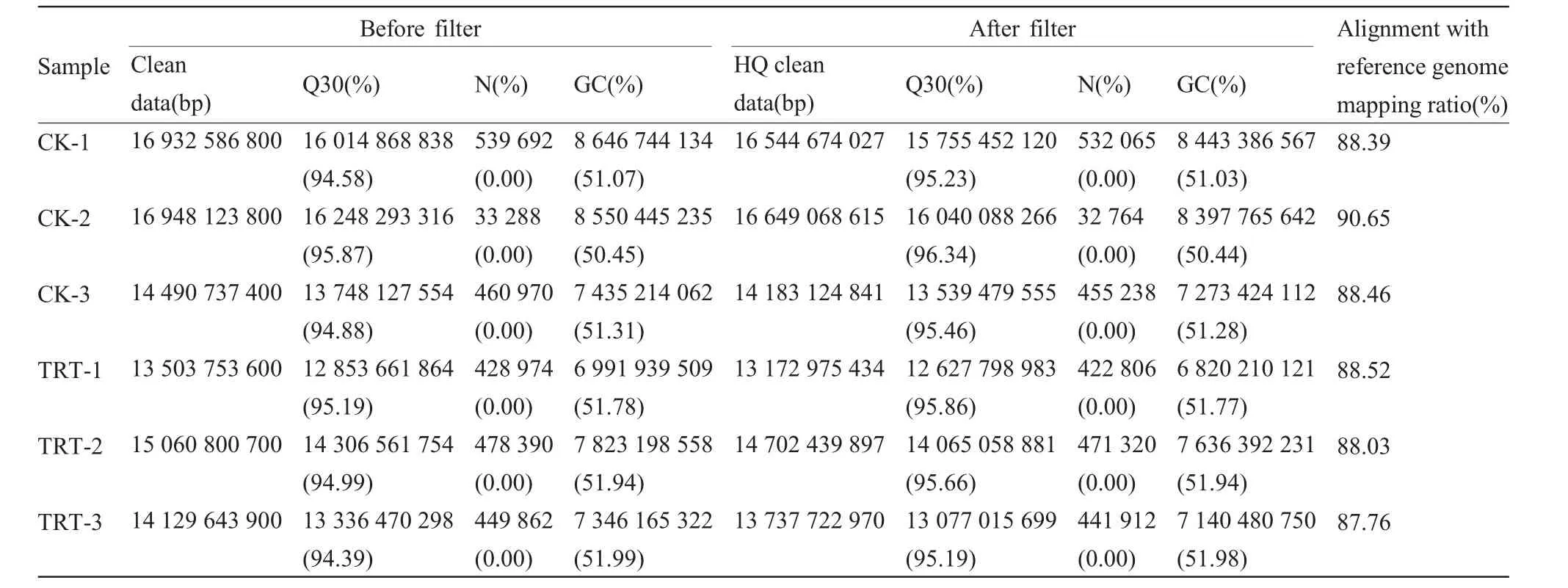
表2 过滤前后碱基信息及比对分析统计表Tab.2 Base information before and after filtering and comparison analysis statistics table
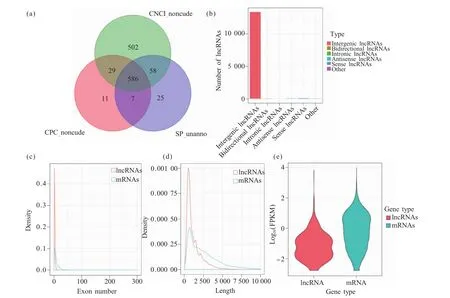
图1 新lncRNAs转录本和数据质量分析Fig.1 New lncRNAs transcript and data quality analysis
2.5 lncRNAs顺式调控靶基因的预测
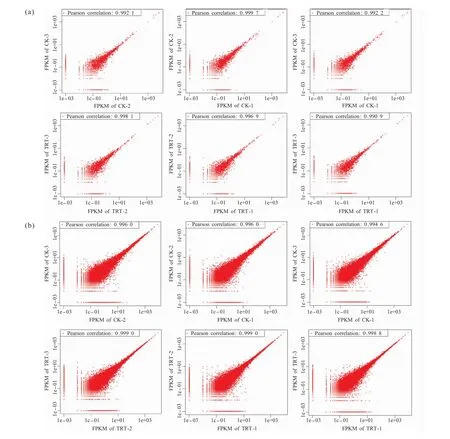
图2 成对重复性检验Fig.2 Pairwise repeatability test
以lncRNAs的上游或者下游10 kb为界定线,我们预测到了108个差异表达lncRNAs具有靶基因。靶基因GO富集分析显示,其显著富集的条目有32个,分别为参与的生物过程16个,细胞组分7个,分子功能9个。参与的生物过程中富集的条目有代谢过程、发育过程、生物调节、细胞过程等,其中细胞过程富集基因最多,有66个。细胞组分中富集的条目有细胞器、细胞器部分、细胞部分、细胞等,其中细胞部分和细胞器富集基因最多,都有66个。分子功能中富集的条目有核酸结合转录因子活性、结合、结构分子活动、催化活性,其中结合功能富集基因最多,有54个(图4a)。KEGG分析结果显示,靶基因富集在了66条通路中。根据KEGG气泡图可知,富集显著性排名前20的通路为:安非他明成瘾、胰高血糖素信号途径、醛固酮合成与分泌、胰岛素分泌、多巴胺能突触、心肌细胞肾上腺素能信号、可卡因成瘾、甲状腺激素合成、HTLV-I感染、单纯疱疹感染、低氧诱导因子-1(hypoxia inducible factor-1,HIF-1)信号通路、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路、哺乳动物寿命调节途径、TNF信号途径、雌激素信号途径、炎症介质对瞬时受体电位(transient receptor potential,TRP)通道的调节、肠免疫网络用于免疫球蛋白 A(immunoglobulin A,IgA)的产生、原发性免疫缺陷、乙型肝炎和甲型流感(图4b)。
2.6 lncRNAs共表达靶基因的预测
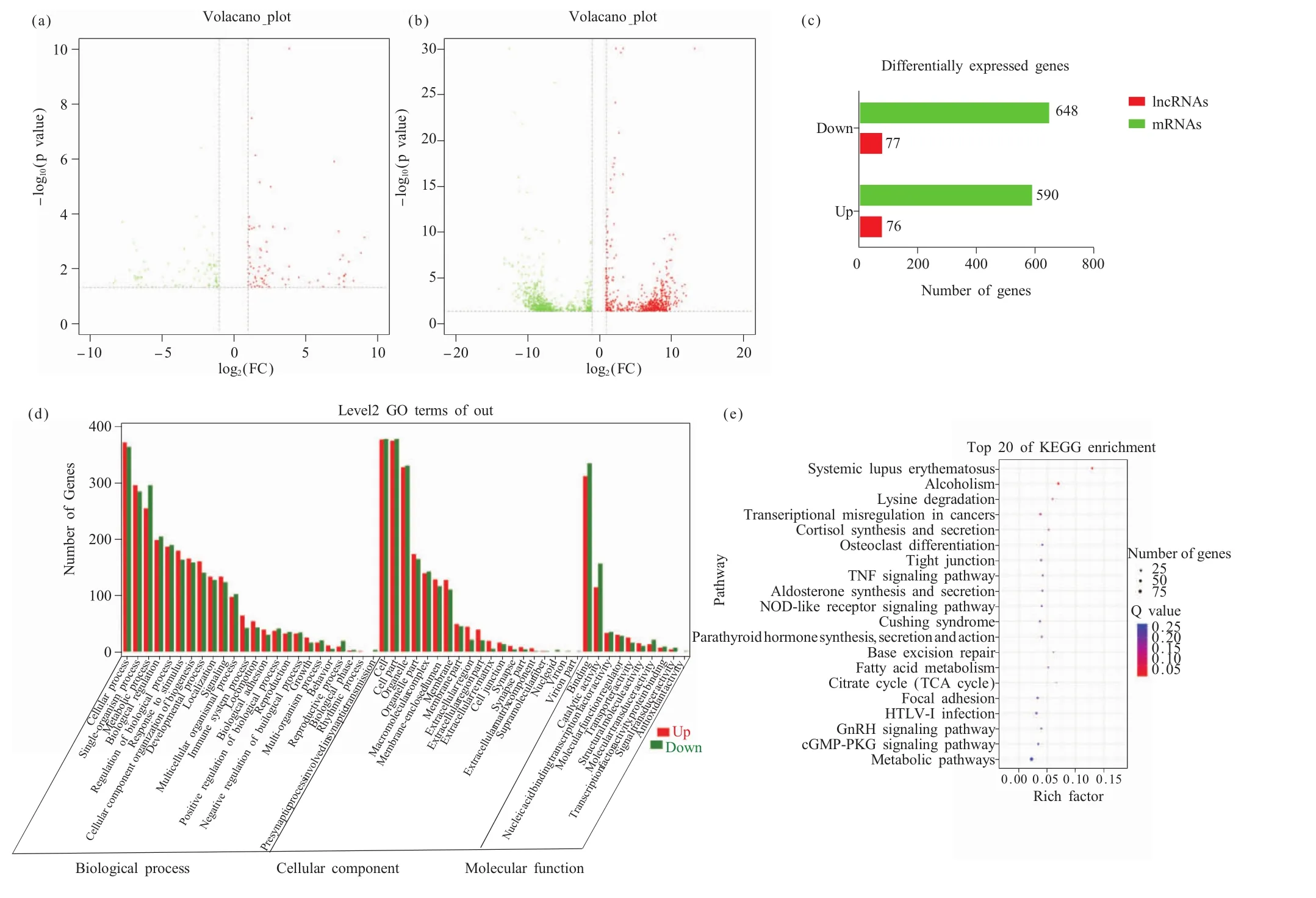
图3 组间差异表达基因分析Fig.3 Analysis of differentially expressed genes between groups
lncRNAs共表达靶基因的GO分析结果显示(图5a),预测到的靶基因富集在了33个条目中。参与生物过程本体中的条目有单生物过程、细胞过程、代谢过程、对刺激的反应等,其中细胞过程富集基因最多,有2 244个。在分子功能中的主要条目有结合、分子功能调节剂、核酸结合转录因子活性、结构分子活性、转录因子活性、蛋白质结合等,其中结合富集基因最多,有1 491个。在细胞组分中的主要条目有细胞外区域、细胞外基质、细胞、细胞器、细胞器部分等,其中在细胞部分富集基因最多,有2113个。
KEGG分析结果显示,共表达基因主要参与290条代谢通路,其中和脂肪分化相关的信号通路有13条,包括cAMP信号通路、过氧化物酶体增殖物激活型受体(peroxisome proliferator activated-receptors,PPARs)信号通路、WNT信号通路、胰岛素分泌、脂肪酸代谢、叉头转录因子(the forkhead box O,FOXO)信号通路、脂肪酸生物合成、脂肪细胞脂解的调节、胰岛素抵抗、MAPK信号通路、胰岛素信号途径、Janus激酶信号转导与转录激活子(the Janus kinasesignal transducer and activator of tran-ions,JAK-STAT)信号通路、磷脂酰肌醇3激酶-蛋白激酶B(phosphatidylinositol 3'-OH kinase/protein kinase B,PI3K-AKT)信号通路。根据Q value从小到大排序(图5b),前20富集的通路有:神经活性配体-受体相互作用、cAMP、钙信号通路、细胞粘附分子(cell adhesion molecule,CAMs)、吗啡成瘾、胆汁分泌、胃酸分泌、安非他明成瘾、蛋白质消化吸收、炎症介质对TRP通道的调节、癌症中的蛋白多糖、谷氨酸能突触、醛固酮合成与分泌、胰腺分泌、可卡因成瘾、癌症中的microRNAs、醛固酮调节的钠再吸收、心肌细胞肾上腺素能信号转导、5-羟色胺能突触、细胞因子-细胞因子-受体相互作用。
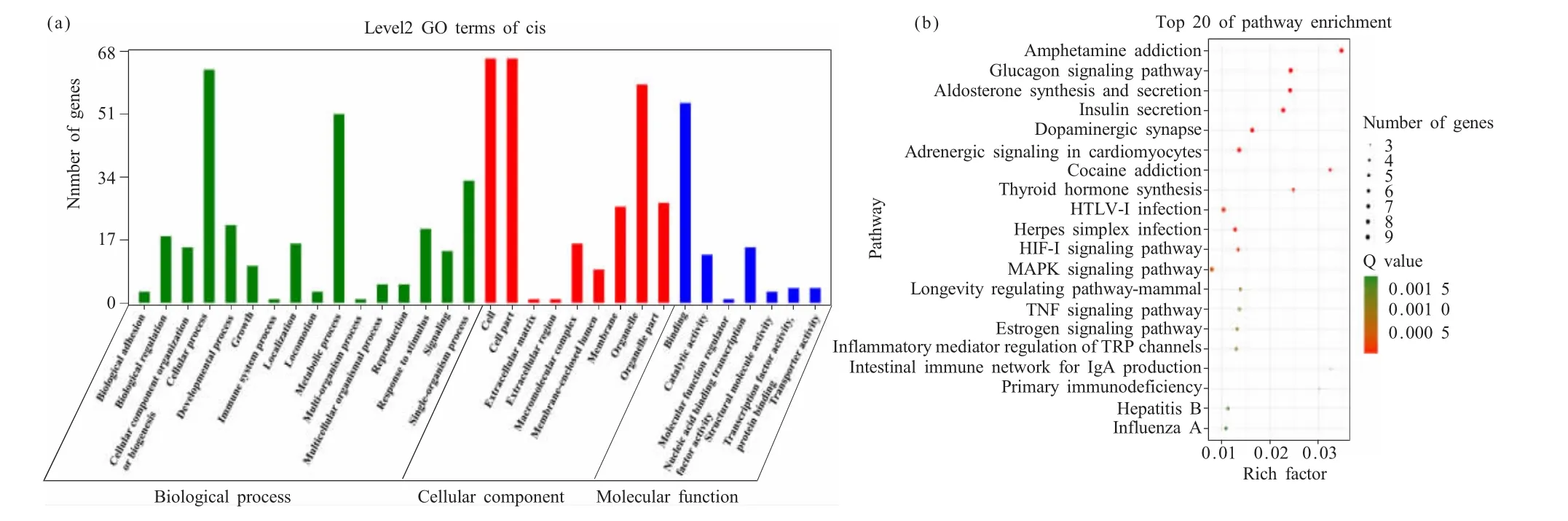
图4 lncRNAs顺式调控靶基因的预测Fig.4 Prediction of lncRNAs cis-regulated target genes
在共表达结果分析的基础上,为了进一步获得目的lncRNAs及其靶向基因,我们将DElncRNAs和DEGs之间符合PCC>0.7或PCC<-0.7且P<0.05的两者定义为具有关系,并使用Cytoscape软件构建了网络互作图。根据研究结果(图6),我们共筛选出了31个差异lncRNAs,它们与134个DEGs相关,其中 1个 DElncRNA(TCONS_00027033)最多可靶向21个 DEGs,1个 DEG(ENSMUST00000114960)最多可与12个DElncRNAs共表达。
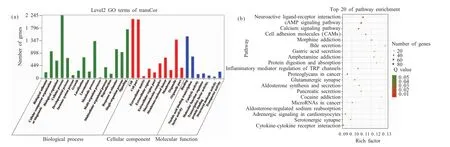
图5 lncRNAs共表达靶基因的预测Fig.5 Prediction of lncRNAs co-expression target genes
2.7 qRT-PCR验证结果
为了验证测序数据的准确性,本研究使用qRTPCR对随机选择的3个lncRNAs和3个mRNAs进行定量验证。测序结果中FC值>1或<1分别代表基因的上调或下调和表达量的升高或降低。结果表明qRT-PCR表达趋势与测序结果一致,lncRNAs(ENSMUST00000152125、ENSMUST00000191042、TCONS_00033926)及 mRNAs(ENSMUST000000109-74、ENSMUST00000031327)在荧光定量PCR与测序结果中都上调,而mRNA(ENSMUST00000165806)下调(图7)。这证明了本次测序结果的可靠性。
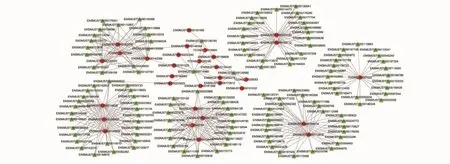
图6 DElncRNAs与DEGs的共表达网络互作图Fig.6 Co-expression network mapping of DElncRNAs and DEGs
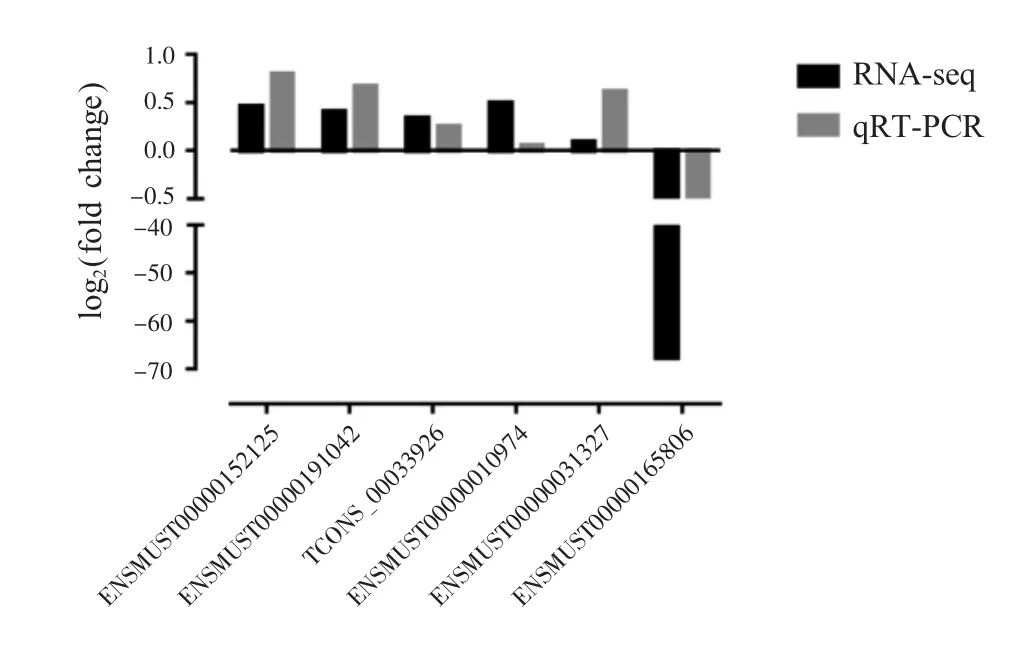
图7 差异表达lncRNAs的验证Fig.7 Validation of differentially expressed lncRNAs
3 讨论
肥胖及其带来的代谢类疾病已受到科学界的广泛关注。目前应用于治疗肥胖的方法包括饮食干预、运动治疗、药物治疗及外科手术等[23],但上述疗法具有一定的局限性和副作用[24]。近年来的研究表明,BAT激活增加能量消耗可作为防治肥胖症的潜在靶点,通过激活BAT增加能量消耗是一种安全预防和控制肥胖的方法[25]。在模式动物中,增加和移植BAT能促进脂肪代谢,减轻体重,并改善全身胰岛素的敏感性[7,26]。我们前期的研究表明,APS可以促进C3H10T1/2细胞的成脂分化,并提高其产热能力。本研究旨在阐明APS改变了C3H10T1/2分化中lncRNA的表达谱,为从lncRNAs的角度揭示APS促进BAT细胞分化的分子机制提供科学依据。
RNA-seq技术的广泛使用改变了我们对真核细胞转录组程度和复杂性的观点。与其他方法相比,RNA-seq提供了更精确的转录物水平和转录物之间的联系[27],已被广泛地应用于 lncRNAs、micoRNAs和mRNAs等的研究中。lncRNAs的平均表达量、长度、外显子个数均要小于蛋白编码基因[28],这与我们的测序结果一致,进一步说明了本次测序结果的准确性。目前在lncRNAs顺式靶基因预测中,选用的标准是lncRNAs上下游300 kb之内的基因定义为相关,但多选择100 kb[29-30]。本次试验我们采用了更加严苛的要求,筛选了上下游10 kb之内的基因定义为相关,这增大了获得与BAT分化相关靶基因的概率。DElncRNAs的顺式及共表达预测到的靶基因GO分析中,我们发现顺式及共表达靶基因在分子功能本体中富集的条目保持着一致,这些条目包括核酸结合转录因子活性、转录因子活性、蛋白质结合等。这进一步证明了lncRNAs在基因组中具有调节基因表达的作用。
在对预测到的DElncRNAs顺式调控靶基因进行KEGG分析后发现,靶基因在胰高血糖素信号途径中富集显著。胰高血糖素是一种主要来源于胰岛α细胞的激素,在生理上通过独特的受体来维持葡萄糖的稳态。研究发现,在BAT组织的切片中直接施用胰高血糖素会增加氧气消耗和游离脂肪酸的释放[31]。同时,胰高血糖素对BAT细胞的刺激可以诱导热量的产生[32]。低温环境下发现,敲除胰高血糖素基因的小鼠其BAT中的生热标记(例如:Ucp1、Dio2和Ppargc1α)表达也在降低[33]。上述结果都证明了胰高血糖素对棕色脂肪细胞的代谢和分化具有重要的作用。外源性的APS可能通过改变相关lncRNAs的表达,调控多个处于胰高血糖素信号途径的基因,从而影响BAT细胞的分化。在富集显著程度居于前20的通路中,我们还发现胰岛素分泌[34]、MAPK信号通路[35]、TNF等信号通路[36]都对BAT分化具有重要作用。在本研究结果中,前两者信号通路中分别各有8个预测到的靶基因富集,后者有5个基因富集。综上可知,DElncRNAs顺式调控的靶基因富集于多条关于BAT分化的信号通路上的结果揭示了APS可能通过改变lncRNAs的表达来调控BAT的分化。
共表达靶基因KEGG通路分析结果表明,多个基因显著地富集在了cAMP信号通路上。cAMP信号通路是一条调节脂肪细胞分化的经典途径,体内的多种激素和细胞因子都是通过此途径来调节脂质代谢和脂肪细胞分化的。细胞中cAMP的含量能决定蛋白激酶 A(protein kinase A,PKA)的活性,其能通过磷酸化2种脂质代谢重要的酶蛋白周脂素(perilipin A)和激素敏感脂酶(hormone-sensitive triglyceride lipase,HSL)来抑制脂肪细胞的分化[37]。同时细胞中cAMP浓度升高也可使磷酸二脂酶(phosphodiesterase,PDE)磷酸化从而被激活。PDE有14个亚基,分属于7个家族[38],其中脂肪细胞中的PDE3B的活性与脂肪细胞分化呈正相关[39]。本研究还发现了多个靶基因富集在脂肪代谢的途径,包括14个脂肪酸代谢,6个脂肪酸生物合成、6个不饱和脂肪酸的生物合成和6个脂肪酸降解,同时发现靶基因富集在了 PPAR信号通路、WNT信号通路[40]、JAKSTAT信号通路、PI3K-AKT信号通路等与脂肪分化的相关的通路中。这些结果进一步表明,添加APS导致的lncRNA表达谱变化对于BAT分化具有重要影响,且这种影响涉及到多个方面,并突出地显示了APS可能主要通过影响cAMP信号通路来加速BAT细胞的分化。
在共表达网络互作图结果的基础上,我们尝试采用FDR<0.05且|log2FC|>1作为筛选条件进一步缩小差异lncRNAs的范围来获取目的lncRNAs。筛选共得到了10个DElncRNAs。在这10个lncRNAs中,我们聚焦了1个显著下调的lncRNA(ENSMUST00000199606),其共表达分析预测靶向基因为ENSMUST00000209061,在对照组和处理组中表达呈显著差异。ENSMUST00000209061基因编码的是具有CCCH型RNA结合结构域且折叠成特殊的指状结构的 RNA结合蛋白锌指蛋白 36(tristetraprolin,ZFP36),其功能主要是通过与富含腺嘌呤和尿苷的mRNA 3'端结合来降解mRNA[41]。许多研究表明,ZFP36与脂肪分化及肥胖形成有关,并将其作为治疗代谢疾病和肥胖的潜在靶点。在3T3-L1前脂肪细胞中,ZFP36能通过与丝裂原活化蛋白激酶磷酸酶 -1(mitogen-activated protein kinase phosphatase-1,MKP-1)mRNA结合来限制其表达,从而抑制脂肪细胞的分化[42]。与正常肥胖者相比,患有代谢综合症的肥胖者内脏脂肪中的ZFP36的蛋白和mRNA水平降低了4~5倍,且ZFP36基因位于19q13.1长臂,是个与代谢综合征相关的区域[43]。有研究在人体中也发现ZFP36基因在网膜脂肪组织中的表达能预防胰岛素抵抗和糖尿病[43]。更有趣的是,ZFP36包含许多丝氨酸和苏氨酸,从而具备广泛磷酸化的潜力,然而磷酸酶处理仅部分改变了蛋白质的迁移率,表明存在其他蛋白质修饰或与其他物质的结合阻止了磷酸酶功能[44]。而lncRNAs很重要的功能就是对于蛋白质的修饰,故我们猜想是lncRNA(ENSMUST0-0000199606)修饰了ZFP36,从而在脂肪分化和肥胖中起到重要作用。然而其真实的作用及具体的作用机制还需要进一步研究证明。
综上所述,本研究结果揭示了APS能影响C3H-10T1/2细胞棕色脂肪化过程中lncRNAs的表达。靶基因预测显示,APS可能主要通过DElncRNAs来影响包括胰高血糖素、cAMP等信号通路中的基因相对表达来促进BAT细胞分化,同时在进一步筛选中我们发现了差异lncRNA与ZFP36的关系。这些结果为进一步研究APS对BAT细胞分化的调控作用提供了科学依据。
猜你喜欢
清华金融评论(2022年4期)2022-04-13
安徽农业科学(2022年1期)2022-02-14
国际放射医学核医学杂志(2021年10期)2021-02-28
昆明医科大学学报(2020年12期)2021-01-26
房地产导刊(2020年7期)2020-08-24
神州·下旬刊(2019年1期)2019-02-11
中国神经再生研究(英文版)(2017年10期)2017-11-08
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
中国药理学通报(2014年2期)2014-05-09
