
KLF2在肺动脉高压大鼠肺组织中的表达及机制探讨*
2020-09-09 06:49尚粉青叶玉兰
陕西医学杂志 2020年8期
王 蕊,尚粉青,叶玉兰,郭 瑄
1.西安高新医院心内科(西安710075);2.西安市第一医院心内科(西安710002)
肺动脉高压(Pulmonary hypertension,PAH)发病隐匿,诊疗棘手,预后差。近年来研究表明,肺血管内皮细胞功能紊乱是导致PAH肺血管异常收缩、肺血管重构和肺部血栓形成的主要原因[1]。Kruppel样转录因子2(Kruppel-like factor2,KLF2)在肺组织高度表达,近来研究表明其在调节内皮细胞生物活性中起重要作用[2],因此我们预测其在PAH发生过程中可能起重要作用。本研究通过腹腔注射野百合碱(Monocrotaline,MCT)建立大鼠PAH模型,观察KLF2及其相关因子表达变化,以期为进一步研究PAH干预措施提供理论依据。
材料与方法
1 实验动物及材料 清洁级雄性SD大鼠24只,8~10周龄,体重(200±10)g,购于西安交通大学动物研究中心,饲养于西安交通大学医学院心血管研究中心动物房;动物房为清洁级,动物的饲养条件:温度(22±2)℃,标准12 h光照12 h黑暗节律,普通专用大鼠合成饲料喂养和饮水。野百合碱由购自Sigma公司。PCR引物由擎科生物技术有限公司合成。β-actin抗体购自Santa Cruz公司;eNOS抗体、KLF2、ET-1抗体购自美国Cell Signaling公司。NK-κB抗体、VEGFR2抗体购自美国Abcam公司。蛋白酶抑制剂及磷酸酶抑制剂(德国Roche公司),ECL发光液(Vazyme公司,Millipore公司),Trizol 试剂(Invitrogen公司),HiScriptTMqPCR Super Mix逆转录试剂盒(Vazyme公司),PCR DNA聚合酶(Vazyme公司),其余试剂为国产分析纯。实时定量PCR仪及梯度PCR仪(美国Applied Biosystems 公司),WB电泳仪和电泳槽、WB半干转膜槽(BIO-RAD公司),自动洗片机(柯达公司)。
2 动物模型的建立 所有实验大鼠适应性饲养1周,随机将其分为对照组(10只)、PAH模型组(MCT组,14只),均在同等条件下喂养。对照组按60 mg/kg腹腔注射0.9%氯化钠溶液,MCT组按60 mg/kg腹腔一次性注射MCT造模。
3 血流动力学和心室肥厚检测 MCT注射造模4 周后,称量两组大鼠体重 (BW),水合氯醛(5 ml/kg)腹腔注射麻醉,连接压力换能器,将微导管 (PE50) 沿着大鼠颈外静脉插入右心室,此时可见振幅较大的右心室波,并记录右心室收缩压(RVSP)。处死动物后取出心肺组织,去除心房及大血管,分离右心室和左心室以及室间隔,滤纸吸干组织并测量右心室质量(RV)、左心室质量(LV)和室间隔质量(S),计算右心室肥厚指数(RVHI)=右心室RV/(左心室LV+室间隔S)、右心室质量指数(RVMI) =RV/BW(mg/g)。肺组织标本一部分组织分成1 cm×1 cm×1 cm大小迅速存放于液氮中,后于-80 ℃冻存,用于分子生物学研究。剩余部分用4%多聚甲醛溶液固定。
4 血清各项指标测定 使用ELISA试剂盒测定血清中白介素-1β(IL-1β)、白介素-6(IL-6)及内皮素-1(ET-1)水平变化。采用硝酸还原酶法测定血清一氧化氮(NO)含量。
5 肺组织各项指标测定 ①取肺组织50 mg,剪碎,加入Trizol 1 ml超声匀浆,提取组织RNA,反转录-聚合酶链反应(RT-PCR)方法测定肺组织中KLF2及核因子-κB(NK-κB)、血管内皮生长因子受体2(VEGFR2)、内皮素-1(ET-1)、一氧化氮合酶(eNOS)各因子的表达变化。PCR引物序列见表1。②取肺组织80 mg/只,剪碎组织,加入蛋白裂解液约300 ml,匀浆机匀浆,3次,每次30 s;液氮冻融3次,4℃,12 000 r/min,离心15 min,取上清,ABC法测定蛋白浓度;Western blot方法测定肺组织中上述各因子的蛋白表达的变化。

表1 RT-PCR引物列

结 果
1 一般情况观察及基本数据 MCT组在腹腔注射第2周开始出现毛发枯糙、喘息、食量减少,第25、26天分别有1只大鼠死亡,实验结束时对照组无死亡。MCT组大鼠RVSP较正常对照组显著增高(P<0.01),提示PAH模型制备成功(图1A);正常对照组大鼠右心室形态正常,RVHI为(23.7±2.1)%,MCT组大鼠RVHI较正常对照组显著增高(33.2±2.2)%(P<0.01)(图1B);正常对照组大鼠RVMI为(0.81±0.15)%,MCT组大鼠RVMI较正常对照组显著增高(1.25±0.24)%(P<0.01)(图1C)。

注:与对照组相比,**P<0.01
2 两组大鼠肺组织中KLF2及NK-κB、VEGFR2、ET-1、eNOS各因子表达比较 RT-PCR结果提示,MCT组大鼠肺组织中KLF2及eNOS的mRNA表达水平较对照组显著降低;而NK-κB、VEGFR2及ET-1的mRNA表达水平较对照组显著增高。Western blot结果提示,MCT组大鼠肺组织中KLF2及eNOS的蛋白表达水平较对照组显著降低;而NK-κB、VEGFR2及ET-1的蛋白表达水平较对照组显著增高。见图2、3。
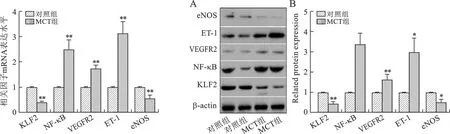
注:与对照组相比,**P<0.01 注:与对照组相比,*P<0.05,**P<0.01
3 两组大鼠血清中IL-1β、IL-6及ET-1、NO水平测定结果 MCT组大鼠血清中IL-1β、IL-6及ET-1水平较对照组显著升高,而NO水平较对照组显著降低,见表2。
表2 两组大鼠血清各因子表达情况
4 大鼠肺组织表达KLF2情况与血清各因子的相关性分析 见图4。
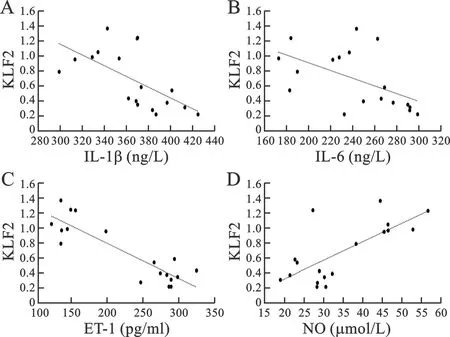
图4 大鼠肺组织KLF2与血清各因子相关性
Spearman相关性分析结果显示:KLF2与血清IL-1β(r=-0.617,P=0.0006)(图4A)、IL-6(r=-0.539,P=0.021)(图4B)及ET-1(r=-0.881,P=0.006)(图4C)呈显著负相关,而与血清NO含量呈显著正相关(r=0.721,P=0.001)(图4D)。
讨 论
肺动脉高压指肺动脉压异常升高的一种病理生理状态,其发病机制复杂,尚未完全明确。目前治疗主要集中在抑制肺血管平滑肌细胞增殖及肺血管收缩,波生坦作为一种ET-1受体拮抗剂应用于临床取得了可喜的疗效[3];西地那非为磷酸二酯酶V选择性抑制剂,能增强NO释放;外源性前列环素类似物,主要通过扩张肺动脉血管床[4]。亦有多种中药显示出一定的控制肺动脉高压的作用[5-6]。上述药物虽然可以改善生活质量,但是仍不能有效改善预后。因此需要进一步探讨其发病上游更重要、更集中的因素所在,为进一步研究治疗靶点提供理论依据。
目前有观点表明,肺血管内皮细胞功能紊乱是PAH发生发展的基础环节。KLF2作为KLF家族中重要的转录因子,在内皮功能调控中起重要作用。我们推测KLF2在PAH发病过程中可能有以下作用机制。①调节血管活性因子的生成:KLF2可抑制ET-1和ACE的表达,降低缩血管物质的生成[7];同时可直接结合到eNOS基因启动子增加其转录活性[8],效诱导e NOS的表达,使血管NO生成增加。②越来越多的证据表明,炎症反应在肺动脉高压病理改变的形成中发挥关键作用[9-10];如炎症因子IL-1β、IL-6、MCP-1及TNF-a均明显增高[11];KLF2通过抑制炎性因子IL-1β、IL-18等的产生发挥抗炎作用[12]。研究表明[13]NF-κB在肺高压大鼠肺组织表达显著增高,参与肺高压发展过程中氧化应激及炎症反应的调控过程。KLF2可能与抑制NF-κB通路的多个组件相互作用,降低NF-κB的活性,间接发挥抗炎作用[14]。③血管增生重构:KLF2与VEGFR2启动子结合抑制内皮细胞VEGFR2的表达[15],从而有抑制血管生成的作用;另有研究报道KLF2可抑制低氧诱导因子的表达,抑制低氧诱导的血管生成[7]。
本研究结果揭示PAH大鼠肺组织KLF2表达明显减少。前期相关研究[16]亦揭示缺氧诱导的肺动脉高压大鼠肺组织KLF2显著降低,AMPK磷酸化ACE2后可诱导KLF2表达增加从而缓解肺高压进展。肺动脉高压缩血管物质ET-1表达显著增加,而e NOS表达降低,NO生成减少,ET-1与NO均可被KLF2调控其表达,提示KLF2为多种血管活性物质的共同调节因子。同时,PAH大鼠肺组织NF-κB表达显著增加,血清IL-1β表达亦明显增高,提示炎症因子表达明显增高,KLF2可调节NF-κB的 表达及活性,从而对内皮细胞炎症反应过程起到调控作用。由本研究结果可推测,KLF2在PAH发病发展过程中可能发挥关键作用,是多种血管活性物质生成的上游调节因素,是炎症反应的上游调控因子,是血管重构的调节因素。因此,针对KLF2来进行PAH的治疗干预措施的研究将是未来的方向之一。
猜你喜欢
中国临床医学影像杂志(2022年7期)2022-12-10
军民两用技术与产品(2022年6期)2022-08-06
临床医学工程(2022年5期)2022-05-19
现代临床医学(2021年2期)2021-03-29
心肺血管病杂志(2020年5期)2021-01-14
心肺血管病杂志(2020年5期)2021-01-14
学习与科普(2019年3期)2019-09-10
廉政瞭望(2018年15期)2018-09-17
大众健康(2016年6期)2016-08-03
民生周刊(2015年6期)2015-03-23
