
金属掺杂TiO2纳米粒子电化学催化合成氨
2020-09-05 04:18段国义陈咏梅孙艳芝万平玉
化学研究 2020年3期
任 远,段国义,陈咏梅,孙艳芝,唐 阳,万平玉
(北京化工大学 化学学院,北京 100029)
氨(NH3)是生产化肥和其他化工产品的重要化工原料之一[1].目前,全球每年氨需求量超过100万吨,其中95%是以Haber-Bosch工艺为基础而生产的[2].Haber-Bosch工艺需要依赖400~500 ℃高温和铁基催化剂,其中氢气需要从甲烷重整或水煤气而来,而N2的单程转化率只有15%左右,因而该工艺被认为是一个极度依赖化石能源、高碳排放与资本和设备密集的工艺过程[3-5].近年来,研究者们正在积极尝试在常温或常压或用H2O作为氢源合成氨的可能性.若以H2O替代H2,则合成氨反应由原来的自发过程(式1)变为非自发过程(式2),需要借助光能或电能的输入[6-7].
-32.90 kJ·mol-1)
(1)
2N2(g) + 6H2O(l) →
656.30 kJ·mol-1)
(2)
电化学合成氨(EAS)体系的优势在于常温常压下电解H2O直接提供氢源,且来自太阳能、风能或水能的可再生电力满足反应所需的能量[8].理论研究推测,绝大多数温和条件下EAS应遵循缔合机理,即催化剂表面吸附的*N2直接发生加氢反应,直到形成(或接近形成)吸附NH3分子(*NH3)时才断开N≡N键[9].可以看出,选择对N2分子有效吸附从而削弱N≡N键能的催化剂是提高合成氨速率(RNH3)的必要条件.除此之外,有效抑制析氢反应(HER)是提高电流效率(FE)的必经之路[10-11].
已有研究表明EAS体系中高的RNH3和FE往往不可兼得[12-13],主要原因是催化剂表面的N2吸附位点常常同时表现析氢活性,因而需要综合考虑催化剂结构设计并进行电解条件优化,以求达到RNH3和FE的最佳值.TiO2具有化学性质稳定、结构可控的特性,多项研究表明在制备过程中通过金属掺杂可在TiO2晶格内部或表面引入氧空位(OVs),这些金属掺杂TiO2(TiO2-M)在光催化合成氨体系和电化学体系中表现出较好的合成氨催化活性[14-16].然而,这些掺杂金属本身在水溶液电化学体系中也是析氢活性中心.为此,能否从掺杂元素种类筛选和电解条件优化两方面进行调制,而使这些TiO2-M催化剂在提高RNH3与抑制析氢二者之间进行选择和优化,是本文的出发点.
基于溶胶-凝胶法制备了Pd、Mn、Cu等多种过渡金属离子掺杂的TiO2纳米颗粒,并将这些TiO2-M颗粒负载在碳纸上制作阴极(TiO2-M/CP),考察了其在水溶液体系中不同电解条件下合成氨的速率和电流效率.此外,通过线性扫描伏安(LSV)测试研究了TiO2-M/CP的阴极极化行为,借此对掺杂金属种类对RNH3和FE的影响进行了解释.
1 实验部分
1.1 TiO2-M纳米颗粒及其修饰电极的制备与表征
室温下,在含有1.00 mL去离子水和1.00 mL浓盐酸(12 mol·L-1)的40.00 mL无水乙醇中,加入3.00 mL预先配制好的Cu2+溶液,控制Cu2+的加入量为0.35 mmol.搅拌20 min后,搅拌下滴入含有12.00 mL钛酸四丁酯溶液,继续搅拌直至最终形成凝胶.室温下老化24 h后,在100 ℃烘箱中干燥12 h,再在450 ℃马弗炉中煅烧2.5 h后得到粉体.所得到的粉体记作TiO2-Cu.按照相同的流程,以相同体积的Mn2+、Pd2+溶液替代上述Cu2+溶液,金属离子加入量均为0.35 mmol,所得的粉体分别记作TiO2-Mn、TiO2-Pd.此外,用相同体积的去离子水替代上述盐溶液,得到无掺杂TiO2粉体,在本文中作为空白对照样品.
将制备好的粉末在超声振荡下分散到Nafion溶液中,然后将其滴加在碳纸上,干燥后得到碳纸电极,控制催化剂的负载量为(1.80.1) mg·cm-2,记作TiO2-M/CP (M=Cu、Mn、Pd)电极.
粉体及负载在碳纸上的形貌、晶体结构及掺杂元素存在状态通过SEM、XRD和XPS等进行表征.扫描电子显微镜(SEM)图片通过Zeiss SUPRA55扫描电子显微镜(Zeiss,德国)收集,加速电压为20 kV.XRD表征在Ultima IV衍射仪(Rigaku,日本)上进行,使用Cu Kα射线,波长为0.154 06 nm,电压和电流分别为40 kV和150 mA.X射线光电子能谱(XPS)测试通过Escalab 250Xi X射线光谱仪(ThermoFisher,美国)得到,激发源为Al.透射电子显微镜(TEM)图像由JEM-2010透射电子显微镜(JEOL,日本)得到.
1.2 电化学研究
1.2.1 线性扫描伏安(LSV)测试
在室温下,以0.01 mol·L-1K2SO4溶液为电解质,采用三电极体系进行LSV测试.其中,工作电极为TiO2-M/CP,辅助电极为铂电极,参比电极为Ag/AgCl电极,测试仪器为CHI 660E,扫描速率为50 mV·s-1.测量过程中,在工作电极表面附近通入Ar或N2(流速维持60 mL·min-1).
1.2.2 EAS实验
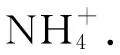

图1 实验装置Fig.1 Schematic diagram of the setup
1.3 产物氨的定量检测
1.4 合成氨速率和电流速率的计算
合成氨速率通过以下公式进行计算:
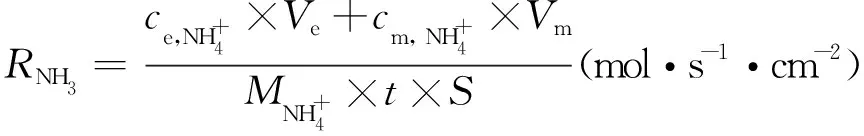
(3)
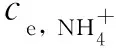

(4)
F是法拉第常数(96 485 C·mol-1),电量的单位为C.
2 结果与讨论
2.1 TiO2-M的表征结果分析
XRD谱图(图2a)表明制备的TiO2及TiO2-M粉体均为纯锐钛矿型(JCPDS 84-1286).此外,TiO2-M的XRD谱图中未发现与金属氧化物(如CuO、MnO和PdO)有关的其他衍射峰,所有样品的(101)晶面衍射峰(2θ=25.5)均无偏移,表明金属掺杂后的TiO2晶格参数和晶胞大小未发生变化.鉴于Cu2+(72 pm)、Mn2+(80 pm)和Pd2+(86 pm)的离子半径均大于 Ti4+(68 pm)的事实[18-19],说明这些金属元素并未进入TiO2的晶格中,而是嵌入TiO2的表面.
TiO2-Cu的Cu 2p XPS图谱(图2b)中,结合能(BE)为933.0 eV和952.8 eV处的双峰分别归属于Cu 2p3/2和Cu 2p1/2,值得注意的是TiO2-Cu中Cu 2p3/2的BE(933.0 eV)高于Cu2O(931.7 eV)[20]但低于CuO(933.9 eV)[21],意味着掺杂Cu原子与两个不饱和O原子相连(-O-Cu-O-),而不是以Cu2O或CuO形式存在[22].通过比较其他掺杂金属的结合能数据(图2c-d和表1)可以获得相似的结果:TiO2-Mn和TiO2-Pd中掺杂Mn和Pd原子分别以-O-Mn-O-和-O-Pd-O-形式存在于TiO2-M表面.
TiO2-Cu样品的TEM照片(图2e)显示原始粒子大小约为18 nm,在HRTEM照片(图4f)中能观察到相邻条纹间距(d)为0.352 nm,对应于锐钛矿TiO2的(101)晶面,与XRD表征结果一致.

图2 (a) TiO2和TiO2-M的XRD谱图;(b) TiO2-Cu在Cu 2p区域的XPS谱图;(c) TiO2-Mn在Mn 2p区域的XPS谱图;(d) TiO2-Pd在Pd 3d区域的XPS谱图;TiO2-Cu的TEM (e)和HRTEM (f)照片Fig.2 (a) XRD patterns of TiO2 and TiO2-M; (b) Cu 2p XPS spectra of TiO2-Cu; (c) Mn 2p XPS spectra of TiO2-Mn; (d) Pd 3d XPS spectra of TiO2-Pd; TEM (e) and HRTEM (f) images of TiO2-Cu
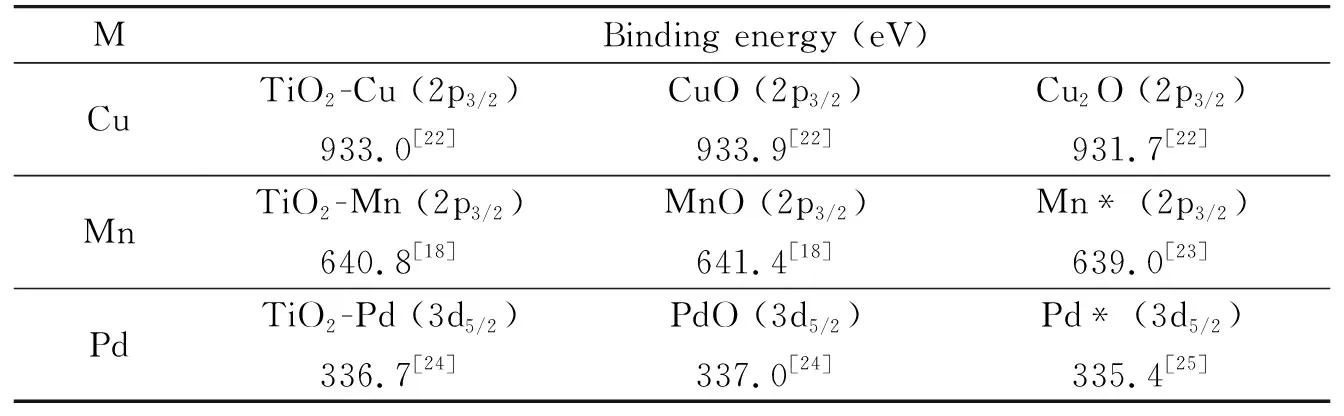
表1 M在TiO2-M和金属氧化物*中的结合能
2.2 TiO2-M/CP的阴极极化行为
分别在Ar和N2饱和的电解液中测试TiO2/CP和TiO2-M/CP的线性扫描伏安测试结果如图3所示.Ar饱和条件下的LSV曲线(黑线)揭示了电极表面HER电化学行为.在阴极电位负移过程中,所有工作电极的阴极电流密度(Jc)不断增大.TiO2-Pd/CP在-1.20 V(vs. Ag/AgCl,下同)时,测得Jc约为7.6 mA·cm-2(图3d),远大于此时TiO2-Cu/CP和TiO2-Mn/CP测得的Jc(图3b和c).这一现象表明TiO2-M/CP表面发生的HER过程是不同的,TiO2-Pd对HER的催化活性最高.N2饱和条件下的LSV曲线如图3中红线所示,此时的LSV曲线是HER行为和EAS行为的叠加结果.显然,在N2或Ar饱和的电解液中,TiO2/CP和TiO2-Pd/CP的LSV曲线基本重合,意味着EAS过程对二者阴极电流的贡献很小.而对TiO2-Cu和TiO2-Mn来说,N2饱和时Jc均在阴极电位负于-0.55 V时明显增大,可与Ar饱和时所得LSV曲线显著区分,显然,增加的Jc是由EAS行为而引起的.在-1.20 V时,TiO2-Cu在N2饱和溶液中测得的Jc为Ar饱和时测得的Jc的3倍,推测TiO2-Cu具有最好的EAS选择性.
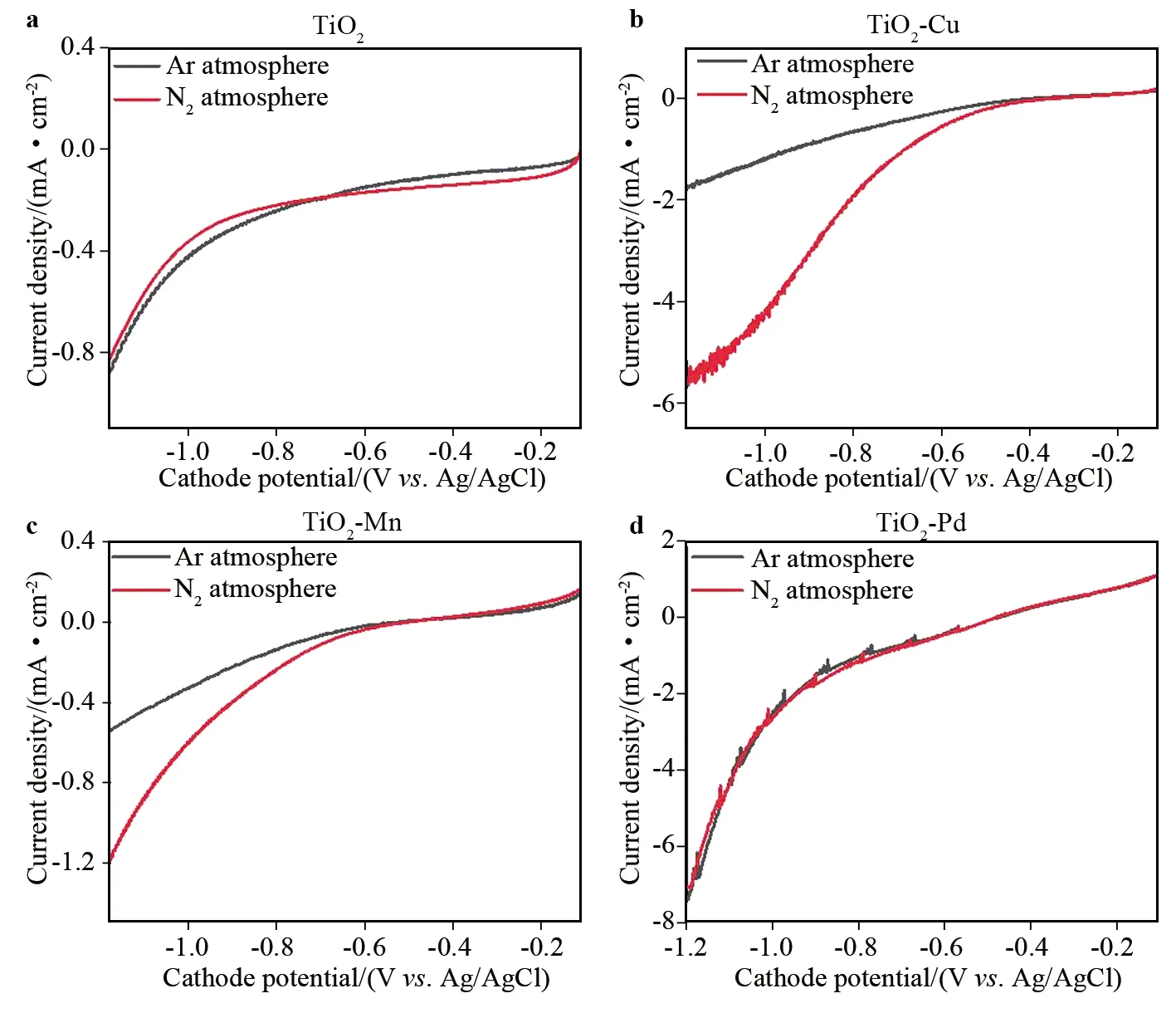
图3 在Ar饱和和N2饱和的电解液中,TiO2/CP和TiO2-M/CP的阴极LSV曲线Fig.3 LSV curves of TiO2/CP and TiO2-M/CP in Ar- and N2-saturated 0.01 mol·L-1 K2SO4 solution
2.3 TiO2-M的电化学合成氨性能及催化机理分析
在相同电解条件下,几种TiO2-M作为催化剂电化学合成氨速率RNH3和电流效率FE如图4a所示.阴极电位为-0.55 V时,三种催化剂的RNH3排序为TiO2-Pd > TiO2-Cu > TiO2-Mn,其中TiO2-Pd的RNH3可达到1.54×10-11mol·s-1·cm-2,而TiO2-Cu和TiO2-Mn的RNH3分别为9.77×10-12mol·s-1·cm-2和8.61×10-12mol·s-1·cm-2;另一方面,实验测得FE排序为TiO2-Cu > TiO2-Mn≫TiO2-Pd,其中TiO2-Cu和TiO2-Mn的FE分别为15.33%和10.76%,但TiO2-Pd的FE仅为0.78%.对此现象的解释如下:已有研究表明TiO2表面掺杂的金属离子处于配位不饱和状态,且其路易斯酸性与金属种类有关,遵循Pd>Cu>Mn的排序[18].也就是说,掺杂的金属离子可以吸附N2分子并减弱NN键能,因而TiO2-Pd表现出最高的合成氨反应速率.然而另一方面这些金属离子同时也是阴极发生析氢反应的催化剂,从图3所示的LSV测定结果可以看出,TiO2-Pd催化析氢的能力也最强,因而其电化学合成氨的电流效率最低.
为了进一步证实析氢副反应对电流效率的影响,本论文以TiO2-Cu为例考察了阴极电位对EAS催化性能的影响.如图4b所示,当阴极电位从-0.55 V负移至-0.75 V时,RNH3从9.77×10-12mol·s-1·cm-2显著提升至1.68×10-11mol·s-1·cm-2, 但FE从15.33%降低至5.51%.随着阴极电位从-0.85 V继续负移至-1.20 V,RNH3和FE都不断降低,这一现象可以通过电生H原子数量的增加来解释:更多的电生H原子应该有利于产物氨的形成;然而,当阴极电位过负(<-0.75 V)时会促进HER,大量生成的H2气泡会占据催化剂表面的活性位点,导致EAS过程被抑制.虽然从LSV曲线(图3)可以看出,TiO2-Cu的选择性在-1.20 V时仍然很好,但实际上此时的RNH3仅为0.6×10-12mol·s-1·cm-2,FE只有0.41%.这是因为LSV测量是一种瞬态分析技术(扫描率为50 mV·s-1),对工作电极近表面的N2分子浓度影响较小;在实际电解过程中,工作电极近表面的N2分子浓度则会迅速下降进而抑制EAS过程,而HER则在-1.20 V时变得更加剧烈.此外,TiO2-Cu比未掺杂的TiO2表现出更高的EAS催化活性(图5)也得到了验证.
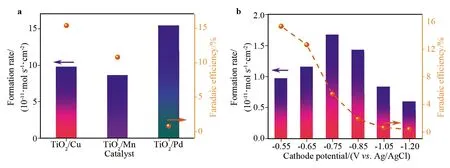
图4 (a) TiO2-M/CPs的EAS催化性能对比图;(b)TiO2-Cu在不同阴极电位下的EAS催化性能Fig.4 (a) NH3 formation rates and FEs of TiO2-M/CP at -0.55 V; (b) NH3 formation rates and FEs of TiO2-Cu/CP at different cathode potentials
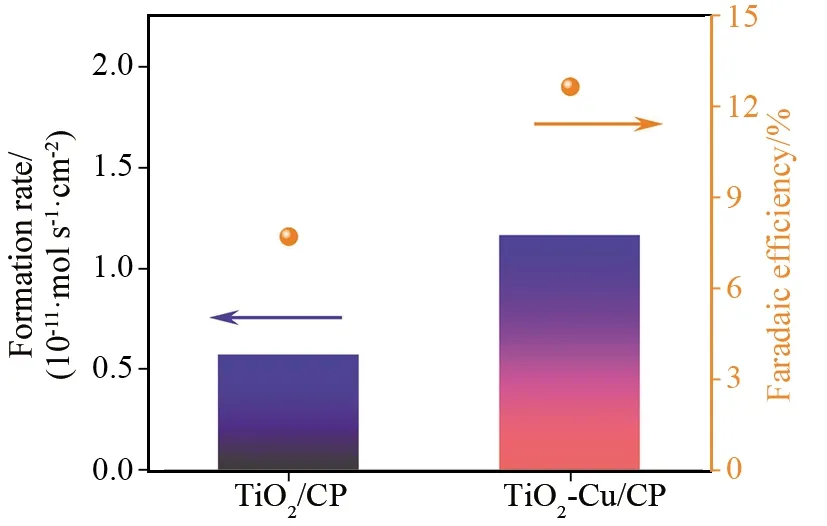
图5 -0.65 V时TiO2-Cu与TiO2的EAS性能对比图Fig.5 NH3 formation rates and FEs of TiO2-Cu/CP and TiO2/CP at -0.65 V for 2 h
3 结论
本研究探讨了不同过渡金属掺杂的TiO2纳米颗粒(TiO2-M)电催化合成氨的催化性能影响因素.研究结果表明掺杂元素种类对EAS反应的RNH3和FE影响显著,TiO2-Pd表现很高的RNH3,但因其析氢催化活性强,因而FE只有0.78%;相反,TiO2-Cu的RNH3中等,但其基本没有HER催化活性,故FE高达15.33%.此外,研究发现阴极电位对TiO2-Cu的EAS催化性能影响如下:当阴极电位从-0.55 V负向移动到-1.20 V时,RNH3呈“钟形”变化,在-0.75 V时达到最大值,为1.68×10-11mol·s-1·cm-2;而FE则不断下降,从15.33%降低至5.51%,这一现象解释为HER活性随电位负移而增强.由此可知,EAS反应要获得高的RNH3和FE,必须精心设计催化剂并优化电解条件以平衡在电极上发生EAS过程和HER过程.
猜你喜欢
中国化肥信息(2022年3期)2023-01-05
中国化肥信息(2022年4期)2023-01-02
中国粉体技术(2022年5期)2022-09-06
轻金属(2022年4期)2022-06-16
中国化肥信息(2022年2期)2022-04-19
中国粉体技术(2022年2期)2022-03-19
世界有色金属(2021年8期)2021-10-31
粉末冶金技术(2021年1期)2021-03-29
粉末冶金技术(2021年1期)2021-03-29
世界有色金属(2020年1期)2020-03-26
