
花生致敏原Ara h 1 的重组表达与纯化
2020-09-01 01:57薛文通
中国食品学报 2020年8期
田 阳 饶 欢 薛文通
(中国农业大学食品科学与营养工程学院 北京100083)
花生过敏是一种发病率高且难以治愈的过敏性疾病,严重时可引发皮炎、哮喘、呼吸困难乃至死亡。 据统计,世界范围内高达1%的儿童对花生过敏[1-2]。花生过敏在美国、加拿大、英国、以色列的儿童发病率分别达到了1.4%,1.7%,1.9%和0.2%。引发花生过敏的蛋白有17 种[4], 其中含量最高(15%)的是Ara h 1[5],研究显示超过95%的中国患者血清可识别该蛋白[6]。 Ara h 1 是同源三聚体,由3 条分子质量均为63.5 ku 的肽链组成[7]。23个致敏表位分布在肽链交联处,使其在热加工、酶解乃至消化过程中仍能保持免疫活性[8]。 研究Ara h 1 的结构及特性,对明确花生致敏机理,生产低致敏花生制品具有重要意义。
如何实现Ara h 1 的有效提取及纯化, 是后续研究的第1 步。 目前较为成熟的提纯方法一般包括硫酸铵盐沉淀、阴离子交换层析、凝胶排阻层析、亲和色谱柱层析等流程[9-10],持续时间较长,步骤繁琐,且成本较高。 Ara h 1 在冗长的提纯过程中容易降解或失活, 层析的过多更会降低目标蛋白的得率。 蛋白的重组表达可有效避免操作流程的繁琐,提高纯度与得率,降低纯化成本。 重组蛋白的基因序列与天然蛋白一致,在结构、功能特性方面与天然蛋白差异较小[11]。 此外,使用重组技术生产的低致敏性蛋白被应用于口服免疫治疗实验中[12],取得了良好的治疗效果。 重组致敏蛋白在研究致敏原结构表征[13]、致敏原检测[14]、致敏机理以及抗敏疫苗[15]的开发等方面正在被愈加广泛的应用。
现有研究表明, 重组Ara h 1(recombinant Ara h 1, rAra h 1)的制备过程仍存在技术难题。 由于原核表达系统与植物源蛋白之间的遗传特性差异较大,所以rAra h 1 在大肠杆菌中的表达程度较低,且易形成包涵体,难以释放在裂解液上清中。蛋白在装配过程中较易错误折叠,形成低分子质量的变体,使得rAra h 1 的纯化变得较为困难。本研究针对以上问题,对基因工程菌诱导过程、菌体裂解条件、蛋白纯化流程进行优化,逐步建立高效的rAra h 1 制备方法,为进一步开展致敏原的结构及生物活性研究提供技术支持。
1 材料和方法
1.1 材料与试剂
pET-32a(+)Ara h 1 重组质粒,由生工生物工程(上海)公司合成。大肠杆菌(Escherichia coli)BL21(DE3)pLysS 感受态细胞,北京全式金生物公司。限制性核酸内切酶SacI/NotI,TAKARA 工程有限公司。 胰蛋白胨、酵母提取物、氯化钠、琼脂糖、氨苄青霉素粉末、甘油,北京均利博生物有限公司。 异丙基硫代半乳糖苷 (isopropyl β-D-1-thiogalactopyranoside,IPTG)、聚乙二醇辛基苯基醚(Triton-100)、 乙二胺四乙酸(Ethylenediaminetetraacetic acid,EDTA)、 十二烷基硫酸钠(Sodium dodecyl sulfate,SDS)、 十二烷基磺酸钠(Sodium laurylsulfonate,SLS)、 咪唑、 二硫苏糖醇 (DLDithiothreitol,DTT)、苯甲基磺酰氟(Phenylmethylsulfonyl fluoride,PMSF)、溶菌酶,北京全式金生物公司。Ni-NTA,Qiagen 公司。SDS-PAGE 分离胶缓冲液(4×,pH 8.8)、SDS-PAGE 浓缩胶缓冲液(4×,pH 6.8)、蛋白上样缓冲液,索莱宝公司。TMB 试剂盒,生工生物工程(上海)公司。 Ara h 1 多克隆抗体,由英国曼彻斯特大学提供。
1.2 仪器与设备
UV-1200 紫外-可见分光光度计,美析仪器有限公司;迷你型蛋白电泳仪,美国Bio-Rad 公司;超声波细胞破碎仪, 宁波新芝生物技术公司;THZ-82 恒温水浴振荡器, 常州国华电器有限公司;SHP-80 细菌培养箱, 林茂科技有限公司;5804R 冷冻离心机, 德国Eppendorf 公司;BHC-1304IIB2 生物安全柜, 苏州安泰空气技术有限公司。
1.3 方法
1.3.1 pET-32a-Ara h 1 重组质粒的制备与鉴定
通过NCBI GENE BANK 数据库查得Ara h 1的基因编号为AF432231.1, 其编码区共有1 895个碱基。在Ara h 1 核酸序列末端插入6×his 标签和终止密码子,序列两端分别添加SacI 和NotI 酶切位点。 重组质粒的构建及检验测序交由生工生物工程(上海)公司完成,保证目标基因的准确插入。
1.3.2 Ara h 1 基因的诱导表达
1)大肠杆菌BL21(DE3)pLysS 的转化与筛选 感受态细胞在冰上融化,每100 μL 感受态细胞中加入20 ng pET-32a-Ara h 1 或空载质粒,轻轻旋转几次混匀内容物,在冰上放置30 min。将混合后的感受态细胞放入预加温至42 ℃的循环水浴中,放置80 s,不可摇动。 快速将离心管转至冰浴,使细胞冷却1~2 min。 将感受态细胞与800 μL LB 培养基混合,37 ℃摇床温育45 min, 使细胞复苏并表达目标基因,温和摇动细胞。分别取转化pET-32a-Ara h 1 和空载质粒的菌液, 均匀涂布于含有50 μg/mL 氨苄青霉素的LB 平板(直径90 mm)上进行菌株筛选。 倒置培养皿,于37 ℃培养12~16 h 后即可观察到有白色的菌落, 即转化子。分别挑取单克隆3 个,在LB 培养基中37 ℃培养过夜。
2)目标基因的诱导表达 在50 mL 离心管中加入10 mL LB 培养基及0.5 mg 氨苄青霉素,按照1/100 的体积比分别加入转化子菌液,37 ℃培养3.5 h。取1 mL 混合的菌液测量OD600,当该值等于0.6 时,留下2 mL 未诱导的菌液作为空白对照, 其余菌液加入300 mmol/L IPTG 后分别在16℃和22 ℃诱导6,20,22 h。 诱导完成后对菌液及空白对照进行4 ℃离心(5 000 g,50 min),留下菌体后用蛋白上样缓冲液进行裂解, 并使用SDSPAGE 鉴定其表达效果。
1.3.3 菌体裂解条件的优化 使用优化的诱导条件大量诱导大肠杆菌BL21 (DE3)pLysS, 在每100 mL 菌液离心得到的菌体中加入10 mL 裂解液(配方见表1),将裂解液插在冰盒中,使用超声波细胞粉碎仪裂解细胞,释放蛋白,直至裂解液变得澄清透亮。 该过程保持低温, 以免蛋白受热变性。 将裂解液4 ℃离心(10 000 g,40 min)后,分别取上清和沉淀制备成电泳样品,在SDS-PAGE 中检测裂解后的目标蛋白表达情况。
1.3.4 重组蛋白rAra h 1 的纯化
1)Ni-NTA beads 预洗 取400 μL Ni-NTA,置于15 mL 尖头离心管中, 离心弃保存液。 加入10 mL 裂解液使其处于稳定缓冲体系,快洗2 次,慢洗1 次(快洗:上、下轻柔颠倒混匀几次,离心力为400 g,4 ℃离心2 min,弃上清;慢洗:旋转培养10 min,离心,离心条件同快洗)。
2)蛋白与Ni-NTA beads 孵育 在预洗过的Ni-NTA beads 中加入10 mL 蛋白上清液, 分别4℃旋转培养40 min、2 h、3 h,4 ℃离心(10 000 g,20 min),弃上清。 使用洗液(20 mmol/L Tris-HCl,20 mmol/L 咪 唑,300 mmol/L NaCl,0.1 mmol/L ED-TA,1 mmol/L DTT,0.4% Triton-100,2%丙三醇,pH 7.4)将吸附蛋白后的Ni-NTA 快洗2 次,慢洗1 次。 洗完后取出10 μL Ni-NTA,加入蛋白上样缓冲液, 做SDS-PAGE 检测Ni-NTA beads 吸附情况。

表1 6 种裂解液配方Table 1 Composition of six kinds of lysis buffer
3)梯度洗脱 取出300 μL Ni-NTA beads于1.5 mL EP 管中,4 ℃离心弃上清。 加100 μL洗脱液A (20 mmol/L Tris-HCl,50 mmol/L 咪唑,300 mmol/L NaCl,0.1 mmol/L EDTA,1 mmol/L DTT,0.4% Triton-100,10%丙三醇,pH 7.4)洗脱两次。 每次洗脱时4 ℃旋转孵育1 h,4 ℃离心(600 g)2 min, 得上清置于新的1.5 mL EP 管中。后继续在Ni-NTA beads 中加入100 μL 洗脱液B(20 mmol/L Tris-HCl,100 mmol/L 咪唑,300 mmol/L NaCl,0.1 mmol/L EDTA,1 mmol/L DTT,0.4%Triton-100,10% 丙三醇,pH 7.4)洗脱两次,步骤同上。 收集洗脱后的上清液,通过SDS-PAGE 检测洗脱液中蛋白种类。
1.3.5 质谱鉴定纯化蛋白的种类 纯化的蛋白经SDS-PAGE 后割下目标条带送至北京邦非生物科技有限公司进行MALDI TOF/TOF 质谱鉴定。 氨基酸序列比对使用MASCOT (Matrix Sciences,英国)软件系统分析。
1.3.6 免疫印迹测定rAra h 1 免疫活性 参考Rao 等[16]的方法,纯化蛋白经SDS-PAGE 后转膜至PVDF 上(80 mA,300 V,90 min),用含5%脱脂奶粉的TBST 封闭(37 ℃,2 h),洗膜3 次,每次5 min,然后将PVDF 膜与1∶5 000(体积比)稀释的Ara h 1 抗体在4 ℃条件下孵育过夜,洗膜后加入1:5 000(体积比)稀释的HRP 酶标羊抗兔IgG 二抗,37 ℃温育2 h。 洗膜后使用TMB 试剂盒显色。
2 结果与分析
2.1 pET-32a-Ara h 1 重组质粒的鉴定
通过使用限制性内切酶Sacl 和Notl 切割重组质粒,并对其产物进行琼脂糖凝胶电泳,结果见图1。 1 号泳道中出现两条明显的条带,表明基因重组质粒构建时并未插入其它杂质基因。 其中5 900 bp 的条带为pET-32a 载体,1 895 bp 左右的条带为目标基因带, 目标带的分子质量数值符合NCBI 数据库中Ara h 1 蛋白的数据。
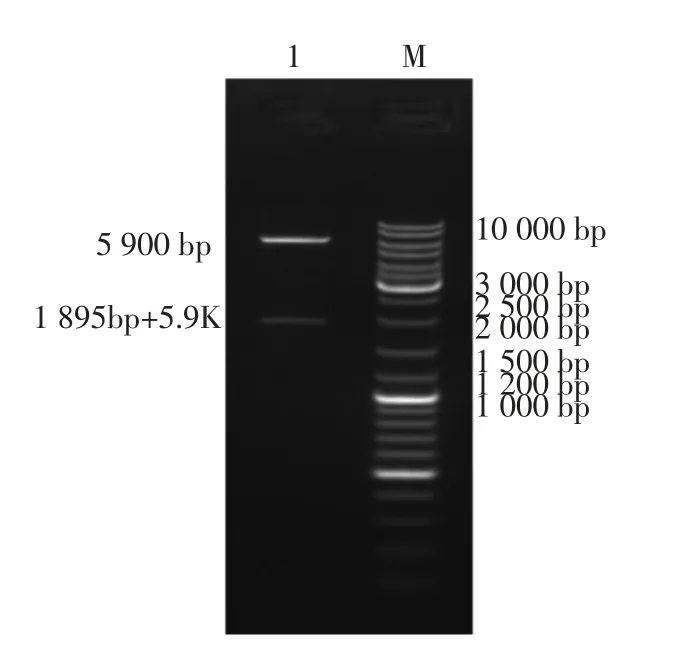
图1 琼脂糖电泳鉴定pet-32a(+)Ara h 1 重组质粒Fig.1 Identification of pet-32a (+)Ara h 1 recombinant plasmid by agarose electrophoresis
精准插入目的基因是评价重组质粒构建成功的重要指标, 也是保证后期目标基因成功表达的重要步骤。 此处通过对重组质粒中的目标基因分子质量、序列进行比对,确认重组质粒的专一性和准确性,为开展下一步试验奠定基础。
2.2 目标蛋白在大肠杆菌中的表达情况鉴定
使用SDS-PAGE 鉴定转化后的大肠杆菌是否成功表达rAra h 1。 如图2a 所示,与泳道1、3、5 相比, 泳道2、4、6 中明显多出1 条分子质量约为110 ku 的条带,该条带亮度较高,是目标基因的表达产物。由此可见,质粒转化后的克隆子均可表达目标基因。与天然Ara h 1 相比,重组Ara h 1 的分子质量增加了46.5 ku。 这主要是由于在pET-32a 标准载体中,启动子之后增加了Trx-tag、His-tag、S-tag 以及9 个酶切位点, 共计1 162 个碱基。多翻译出的序列中包含388 个氨基酸,其分子质量约为46.56 ku, 造成重组蛋白分子质量较高。经课题组试验表明,该蛋白的分子结构及致敏性均与天然Ara h 1 蛋白具有较高的一致性[17],可用于下一步的改性及降敏研究。 相比于泳道2和6,泳道4 中的目标带亮度更高,说明克隆子2号中目标基因的表达效率为最高。 由此选择克隆子2 号扩大培养,大量生产目标蛋白并进行纯化。
根据图2b 中的结果,24 ℃,24 h 诱导后的克隆子2 号,可大量表达目标蛋白(泳道A 中有明显的110 ku 条带), 而菌体裂解液上清中却只有很少的目标蛋白 (泳道B 中110 ku 处几乎没有条带)。 泳道C 中目标带的含量较多,表明大部分目标蛋白被包裹在包涵体内,难以溶解在裂解液中。使用Ni-NTA 对目标蛋白进行吸附和洗脱, 结果如泳道D、E 所示,镍柱无法和裂解液上清中目标蛋白结合, 反而与分子质量约为100 ku 和40 ku等的杂蛋白结合。 这是由于大肠杆菌的诱导条件不适宜,目标蛋白的表达量过少,裂解液上清中的目标蛋白含量显著少于镍柱, 导致镍柱无法有效识别重组蛋白, 转而与杂带结合。 为了提高rAra h 1 的表达效率,使其在裂解液上清中得到释放,对大肠杆菌的诱导条件及菌体裂解条件进行研究。
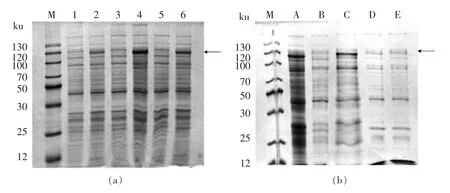
图2 质粒在大肠杆菌BL21(DE3)pLysS 中的表达Fig.2 Expression of plasmid in Escherichia coli BL21(DE3)pLysS
2.3 菌体诱导条件、裂解条件变化对重组蛋白表达量及可溶性的影响
高温短时诱导大肠杆菌虽然可使菌体生长较快,但易造成重组蛋白过度折叠及形成包涵体[18]。由此,选择22,16 ℃分别诱导6,20,22 h。 诱导完后裂解菌体,检测裂解液上清中的重组蛋白含量,结果如图3 所示。图3a 中泳道1、2、3 中110 ku 的蛋白亮度显高于泳道4、5、6, 说明相比于16 ℃和22℃诱导可使重组蛋白获得较高表达量。 此外,泳道2 中的rAra h 1 含量在6 条泳道中为最高,由此确定了22 ℃诱导22 h 为大肠杆菌的最佳诱导条件。
将大量诱导后的菌体进行裂解,结果如图3b所示。相比于泳道A,泳道B 中目标蛋白的表达量较高,说明诱导条件合适。 泳道C 与泳道D 中均存在分子质量为110 ku 的条带,说明优化诱导条件后,目标蛋白形成包涵体的含量减少,重组蛋白已部分溶解在裂解液上清中。
为了增加重组蛋白的可溶性, 使用6 种配方的裂解液对菌体进行裂解,结果如图4 所示。盐溶作用可以破坏蛋白分子表面的水膜, 增加分子表面的电荷,促使蛋白分子与钠离子竞争水分,从而提高溶解效率。 裂解液1、2、3 号中氯化钠浓度逐步上升,而泳道3、5、7 中分子质量为110 ku 的蛋白亮度并未增加, 推测盐溶作用不能使rAra h 1溶解, 因此本试验中将裂解液中的氯化钠浓度确定为较低的200 mmol/L,以免造成蛋白质变性。裂解液4、5、6 号中添加了十二烷基磺酸钠(SLS)作为阴离子表面活性剂,以提高目标蛋白的亲水性。根据图4 显示, 泳道9、11、13 中分子质量为110 ku 的蛋白条带亮度增加,说明添加SLS 可增强目标蛋白的可溶性。与泳道12 相比,泳道14 中目标蛋白的含量明显下降, 说明增加SLS 的浓度可抑制包涵体的形成, 提高重组蛋白在裂解液上清中的表达效率。 据本课题组研究经验,SLS 的浓度过大会抑制his 标签蛋白与Ni-NTA 的结合,因此在裂解液中配方中添加15 mmol/L SLS,在提高蛋白可溶性的同时避免纯化时蛋白与Ni 柱结合的效率过低。
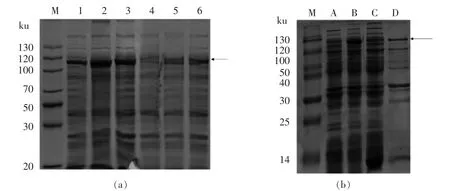
图3 诱导条件对rAra h 1 表达及纯化的影响Fig.3 Effect of inducing conditions on the expression and purification of rAra h 1
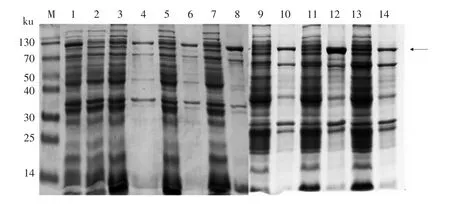
图4 不同配方裂解液中rAra h 1 的溶解度变化Fig.4 The solubility change of rAra h 1 in different lysis buffers
2.4 重组蛋白纯化条件的优化
将裂解液上清与Ni 柱共孵育,通过控制孵育时间的长度,使得杂质蛋白结合较少[19]。 本试验选取40 min,2 h,3 h 3 个时间点终止孵育, 并对Ni柱进行洗脱。使用SDS-PAGE 对目标蛋白与Ni 柱的结合效率、 洗脱结果进行检测, 结果如图5 所示。 图5a 中, 泳道3、5、7 中均出现分子质量为110 ku 的目标蛋白以及90 ku 和22 ku 的两条蛋白杂带,泳道4、6、8 分别是其对应的洗脱结果。与泳道5、7 相比,泳道3 中约90 ku 和约22 ku 的杂蛋白条带亮度较低, 这是由于蛋白液(裂解上清液)与Ni 柱结合时间越短,杂蛋白越难挂柱。本试验最终选择40 min 为Ni 柱结合时间。
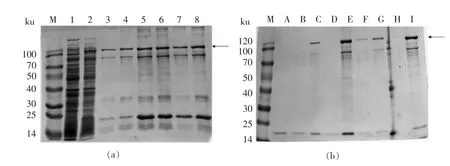
图5 孵育时长和梯度洗脱对rAra h 1 纯化效果的影响Fig.5 The influence of binding time and gradient elution on the purification of rAra h 1
使用梯度咪唑洗脱液洗脱可以控制Ni 柱中结合位点的数量, 从而对目标蛋白的结合效率进行调整,进而获得纯化的重组蛋白。 本试验中,分别采用50 mmol/L 和100 mmol/L 的洗脱液对结合蛋白后的镍柱洗脱2 次, 并对洗脱过程中镍柱结合蛋白的情况做SDS-PAGE 检测。 如图5b 所示,泳道F、G 中杂带含量最低,说明梯度洗脱过程中,使用100 mmol/L 咪唑洗脱蛋白可获得较佳的纯化效果。
2.5 重组蛋白的鉴定与免疫性评价
使用SDS-PAGE 检测诱导前、 后菌体表达蛋白,裂解后上清液,沉淀,结合蛋白后的镍柱以及100 mmol/L 咪唑洗脱液中的蛋白,结果如图6a 所示。泳道5、6、7 中110 ku 的目标蛋白含量高且杂带少,表明rAra h 1 的纯化流程效率较高。 使用质谱鉴定分子质量110 ku 蛋白条带是否与Ara h 1 氨基酸序列相符合, 结果如图6b 所示, 共有9条标志性肽段与Ara h 1 相符,重组蛋白与NCBI数据库中的Ara h 1 氨基酸匹配分值达到426,鉴定该条带即重组Ara h 1 蛋白。
使用western-blot 评价重组蛋白的免疫原性,结果如图6c 所示。 泳道B、C、D、E 中成功表达的重组蛋白均可与anti-Ara h 1 抗体结合, 说明重组蛋白具有与Ara h 1 抗体结合的能力并保持良好的免疫活性。

图6 rAra h 1 纯化结果鉴定及Western-Blot 分析Fig.6 The identification and western-blot analysis of rAra h 1
3 讨论
重组过敏原不仅保留了天然过敏原的抗原性,而且易于制备,方便改性,已广泛应用在过敏原检测和口服免疫治疗疫苗的生产[20-21]。重组蛋白技术可以针对致敏原的线性表位做出定点氨基酸改变,从而靶向改变致敏原的一级结构,获得低致敏性衍生物。 这些低致敏衍生物可在表现较低的IgE 结合能力的同时,保留T 细胞反应活性,因此可用于特异性免疫治疗[22-23]。
随着重组致敏原在过敏病症诊断、 治疗等方面的广泛应用, 重组技术的优化也愈加成为研究热点。目前,已有学者报道Ara h 1、Ara h 2、Ara h 6 的重组制备技术[9,24-26],由于表达载体不同,目标基因序列插入位点不同,所带标签不同等原因,所以各种制备方式存在较大差异, 目标基因表达的效率也各不相同。例如,Gregory 等[27]使用莱茵衣藻制备Ara h 1 和Ara h 2, 虽然表达效率较高,但是重组蛋白的免疫性与天然蛋白相差较大。Lew等[28]对Ara h 2.02 进行密码子优化,使其在大肠杆菌中的表达效率增加,通过亲和层析、蛋白变性和复性等流程对不可溶的重组蛋白进行纯化,操作过程涉及步骤较为繁琐。 闫飞等[9,29]采用RTPCR 获得Ara h 1 基因,构建重组质粒pET-32a-Ara h 1,优化大肠杆菌诱导条件后,利用亲和纯化获得rAra h 1。 本试验前期曾尝试采用该研究条件进行诱导和蛋白纯化, 然而由于质粒构建不同,重组蛋白的可溶性始终较低,对后续纯化工作造成一定困难。 本研究通过对重组Ara h 1 蛋白工艺中的菌体诱导条件、纯化条件等重新优化,得到高效的制备方法, 为后期针对重组Ara h 1 展开结构及生物活性研究奠定了基础。
在制备重组蛋白时, 目标基因的表达很容易形成包涵体,不能溶解在裂解液上清中。这多是因植物源蛋白的遗传信息与原核表达系统相差较大,基因工程菌的诱导条件不适宜所致。若要纯化包涵体中的蛋白,则需要变性、复性等一系列操作[18,30]。 对于致敏蛋白而言,变性与复性可能导致蛋白结构发生改变,进而使过敏原丧失免疫原性。如何促使目标蛋白在裂解液上清中大量表达,是亟需解决的关键问题。研究结果表明,改变裂解液的pH 值,使其远离目标蛋白的等电点,调整裂解液中盐离子的浓度促进重组蛋白的盐溶, 添加阴离子交换剂改善蛋白与水分子的结合效率等措施都可增加重组蛋白的可溶性[31-32]。重组蛋白的制备过程中,诱导条件的确定,菌体裂解液、镍柱平衡液和洗脱液的配方选择尤为重要。 本研究针对以上条件进行研究, 结果表明:300 mmol/L IPTG 于22 ℃诱导菌液22 h 时蛋白表达量最高; 添加15 mmol/L 十二烷基磺酸钠可将蛋白释放在上清液中, 使用含50 mmol/L 和100 mmol/L 咪唑洗脱液分别洗脱2 次和1 次, 可得到纯度较高且免疫原性良好的重组Ara h 1。 rAra h 1 高效制备方法的确定可为之后研究Ara h 1 结构、致敏力变化、致敏机制以及低致敏衍生物的生产提供技术基础。
猜你喜欢
计算机应用与软件(2022年6期)2022-07-12
自然灾害学报(2022年2期)2022-05-10
当代水产(2022年1期)2022-04-26
中国学校体育(2021年10期)2021-04-26
数学大王·低年级(2020年8期)2020-08-14
农产品加工(2018年21期)2018-11-30
中国调味品(2017年2期)2017-03-20
小雪花·初中高分作文(2016年9期)2016-05-14
创新科技(2014年12期)2014-07-27
实验流体力学(2014年6期)2014-07-10
