
基于1H-NMR、31P-NMR的三苯基膦三间磺酸钠定量分析研究
2020-08-21 14:02:34梁春杰孟庆春徐晓婷柴晓飞董昭苹吕印美王岳华蔡颖辉
分析测试学报 2020年8期
梁春杰,孟庆春,徐晓婷,柴晓飞,董昭苹,吕印美,王岳华,蔡颖辉
(黄河三角洲京博化工研究院有限公司,山东 滨州 256500)

图1 TPPTS的分子结构Fig.1 Molecular structure of TPPTS
三苯基膦三间磺酸钠(TPPTS)是一种重要的化工中间体(分子结构式如图1所示),其与金属铑(Rh)配位后可作为羰基合成的催化剂,用于生产比原料烯烃多1个碳原子的醛。与其他催化体系相比,Rh-TPPTS具有选择性好、催化活性高、分离回收易等优点,在正丁醛、正戊醛等醛类化合物的生产工艺中得到广泛应用[1-2]。
TPPTS在制备过程中可能生成多种副产物,给TPPTS的分离分析带来困难。其副产物主要包括以下3类:①苯环上磺酸基的取代位置不在间位;②三苯基膦上磺酸基的取代个数不均一,有可能生成三苯基膦二间磺酸钠以及三苯基膦一间磺酸钠;③TPPTS上的磷原子容易被氧化,生成三苯基氧膦三间磺酸钠(OTPPTS)[3]。由于反应过程中副产物较多,且部分副产物不易分离,至今仍未获得高纯度的TPPTS标准品,难以通过常规液相色谱、气相色谱等方法进行定量分析。而研究表明三苯基膦上的磺酸基位置和数目是影响催化性能的重要因素,且TPPTS被氧化为OTPPTS后其催化性能会受影响[2-4]。因此研究TPPTS合成体系中的副产物种类以及TPPTS在体系中的精确含量,对于后续Rh-TPPTS的催化性能评价与工艺优化具有重要指导意义。早期文献通过元素分析方法来验证TPPTS的产率[5],陈晓华等[6]则通过碘量法测定体系中的三价膦以计算TPPTS含量,但这两种方法不能排除上述①类副产物的影响。截至目前仍未有详细、系统的TPPTS定量分析方法的报道。
近20年来,随着硬件设备的提升以及新型脉冲序列的不断开发,核磁定量逐步在制药、农化、材料科学等领域得到广泛应用[7-12]。与传统气相色谱、高效液相色谱等仪器相比,核磁定量的主要优势在于:①无需待测组分的标准品。②无需对混合体系进行分离,只要待测样品的核磁谱图中有1个信号峰的分离度较好,即可满足定量分析的要求[13]。此外,随着技术的不断进步,核磁定量的准确度也逐步达到甚至超过了传统色谱的定量分析水平[14-17]。
本文采用核磁共振内标法对TPPTS的定量分析进行探究。通过溶剂的选择优化,得到了分辨率高、适于定量分析的一维核磁氢谱;并通过对TPPTS的1H、13C、31P化学位移归属以及纵向弛豫时间(T1)的测定,建立了TPPTS基于1H-NMR、31P-NMR的定量分析方法。两种方法的测定结果一致性高、平行性好,且准确有效。
1 实验部分
1.1 仪器与试剂

1.2 1H-NMR谱峰优化
称取约10 mg TPPTS,分别用DMSO-D6、DMF-D7、D2O-D2溶解,采集1H-NMR,关键采样参数设置如下:脉冲序列:zg30;采样点数:64 K;谱宽:15 ppm;弛豫延迟时间(D1):2 s;采样次数:16;谱中心(O1):6.5 ppm。
1.3 1H、13C、31P化学位移归属
以DMF-D6作为溶剂,分别采集TPPTS的一维1H-NMR、13C-NMR、31P-NMR以及二维1H-1H COSY、1H-13C HSQC、1H-13C HMBC,分别对TPPTS的1H、13C、31P化学位移进行归属。
1.4 纵向弛豫时间(T1)测定
T1的测定均采用t1ir脉冲序列,分别选取10个不同的弛豫恢复时间,具体设置为0.01、0.1、0.25、0.5、1、2、3、4、5、10 s。采集的数据导入Origin 9.0进行曲线拟合。
1.5 核磁内标法定量分析
1.5.231P-NMR定量分析称取约30 mg TPPTS,约10 mg KH2PO4溶于D2O-D2中,实验参数中D1设为35 s,O1为-3.1 ppm。
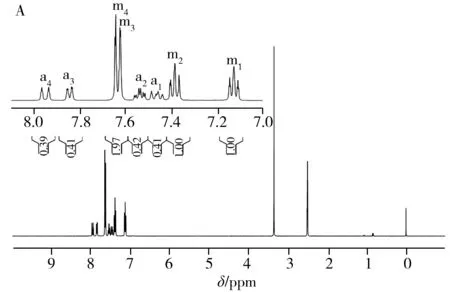
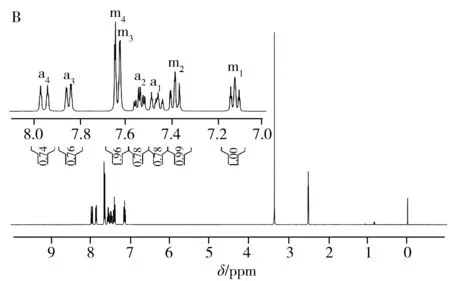
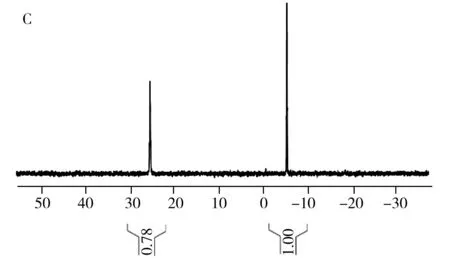
图2 TPPTS在空气中放置前的1H-NMR光谱(A),以及在空气中放置24 h后的1H-NMR(B)与31P-NMR(C)光谱Fig.2 1H-NMR spectrum of TPPTS before(A) exposed to the air, 1H-NMR(B) and 31P-NMR(C) spectra of TPPTS after exposed to the air for 24 h
2 结果与讨论
2.1 1H-NMR中主峰与杂质峰的区分
图2A为DMSO-D6作溶剂时TPPTS的1H-NMR谱图,图中除溶剂峰(δ2.5 ppm)和残余水峰(δ3.3 ppm)外,主要还包含m1、m2、m3、m4四组主峰以及a1、a2、a3、a4其他四组峰。如果把其中1个主峰(如m1)的峰面积定为1,a1~a4四组峰的峰面积均在0.4左右。上述样品在空气中暴露24 h后再次采集一维氢谱,结果如图2B所示,a1~a4四组峰相对于主峰的峰面积较之前变大(由0.40变为0.78左右),说明a1~a4四组峰可能是由于TPPTS被氧化成OTPPTS,苯环上的氢化学位移向低场移动所致;对空气中放置24 h后的样品进行31P-NMR采集,可观察到谱图主要包含2个峰,且低场区较高场区的相对峰面积为0.78(见图2C),基本与图2B氢谱中相对峰面积一致,进一步表明谱图中的杂质峰是由TPPTS上的三价磷原子被氧化成五价磷原子所致。
2.2 氘代试剂的优化
从图2A可以看到,以DMSO-D6作溶剂时,TPPTS的1H-NMR在δ=7.13 ppm处的峰具有较好的分离度,可满足核磁内标法定量的基本要求。但考虑到TPPTS在DMSO-D6中的溶解度很小,在样品称量过程中可能存在较大的相对误差,为此分别采用溶解度较大的D2O-D2以及DMF-D7作溶剂采集了TPPTS的核磁氢谱(图3)。结果显示:以D2O-D2作溶剂时,TPPTS的4组主峰(m1、m2、m3、m4)分离度均不是很好,不适合做定量分析(图3A);而以DMF-D7作溶剂时,TPPTS在δ=7.24、7.40、7.80 ppm处的3组峰(m1、m2、m3)均有较好的分离度,可用于定量分析(图3B)。综合以上分析,最终选择DMF-D7作样品溶剂进行核磁氢谱定量分析。相对于1H-NMR,31P-NMR的谱峰简单且分离度好,因此,本实验选择溶解度较好的D2O-D2作为溶剂,采用磷谱法对TPPTS进行定量分析。

2.3 TPPTS的1H、13C、31P化学位移归属
为了进一步验证上述氢谱中的四组主峰为TPPTS而非其他副产物产生,对TPPTS的1H、13C、31P进行化学位移归属验证,其1H-1H COSY、1H-13C HSQC、1H-13C HMBC信号均可与TPPTS的各原子相匹配(信号归属见图4),由此表明1H-NMR中的四组主峰确实由TPPTS产生,且根据主峰间的相对峰面积可判断定量分析时所选取的信号峰是否存在其他杂质信号干扰(如图3B中m1信号峰若有其他杂质信号重叠,那么m1与m2、m3的峰面积之比将大于1),然后通过选取合适的信号峰以保证定量分析结果的准确性。TPPTS的具体化学位移归属如表1所示。
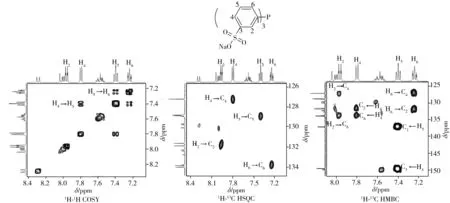
图4 TPPTS的1H-1H COSY、1H-13C HSQC及1H-13C HMBC谱图信号归属Fig.4 Signal assignments of TPPTS by 1H-1H COSY、1H-13C HSQC and 1H-13C HMBC spectra
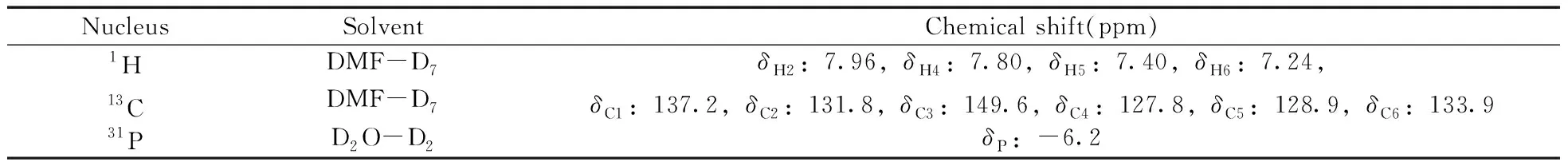
表1 TPPTS的化学位移归属Table 1 Chemical shift assignments of TPPTS
2.4 核磁内标法定量
2.4.1 定量分析原理核磁共振定量的基本原理是在一定条件下体系中的原子核不论所处的化学环境是否相同,单个原子核所产生的峰面积基本相等。因此,只要保证目标化合物中有1个信号峰与其他信号不重叠,再选择1个出峰合适且已知浓度的内标化合物,即可实现目标化合物的定量分析。
假设实验过程中内标化合物的称取质量为m1、相对分子量为M1、质量分数为x1,积分峰面积为S1,积分区包含的原子个数为n1;假设目标化合物的称取质量为m2,相对分子量为M2,质量分数为x2,积分峰面积为S2,积分区包含的原子个数为n2。根据上述定量原理可得:
2.4.21H-NMR定量分析核磁氢谱定量的优势在于灵敏度高,检测时间较短。为了尽可能达到定量原理部分所提到的单个原子核所产生的峰面积相同,需注意以下方面:①多次扫描测量时需确保弛豫延迟时间(D1)大于或等于样品及内标化合物中所检测原子核T1的5倍。②为消除射频脉冲激发偏共振效应,应该把谱中心(O1)置于样品峰及内标峰的中间。③对于有机小分子化合物的定量测试,可在溶解范围内适当增加称样质量,从而减少称量误差。
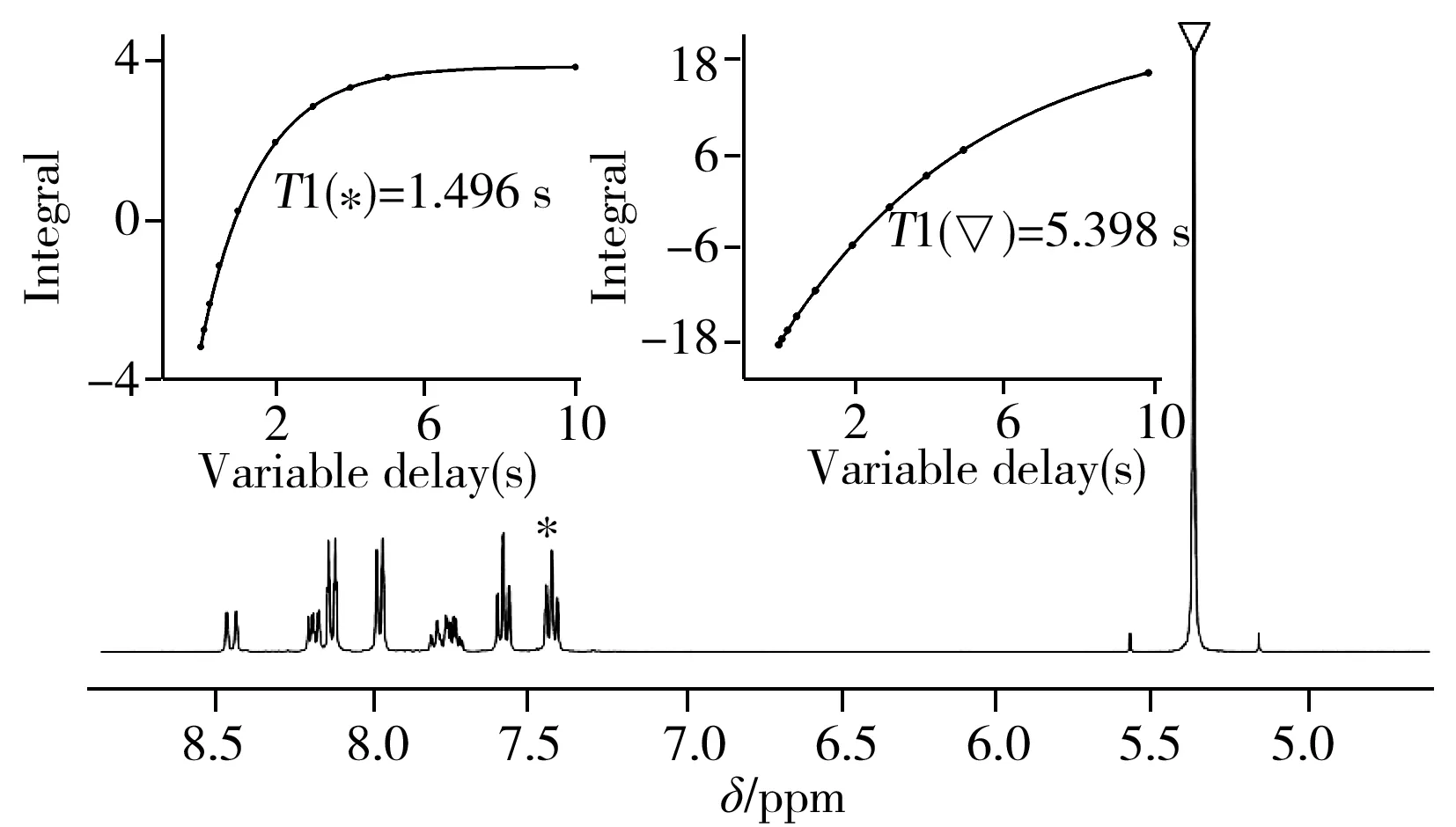
图5 TPPTS和三烷在DMF-D7体系中的1H-NMR谱图Fig.5 1H-NMR spectra of TPPTS and trioxane in DMF-D7insert:T1 fitting curve of the spin nuclear in TPPTS and trioxane(TPPTS和内标峰的纵向弛豫时间T1拟合曲线)

表2 TPPTS的1H-NMR定量分析结果Table 2 Quantitative analysis results of TPPTS using 1H-NMR
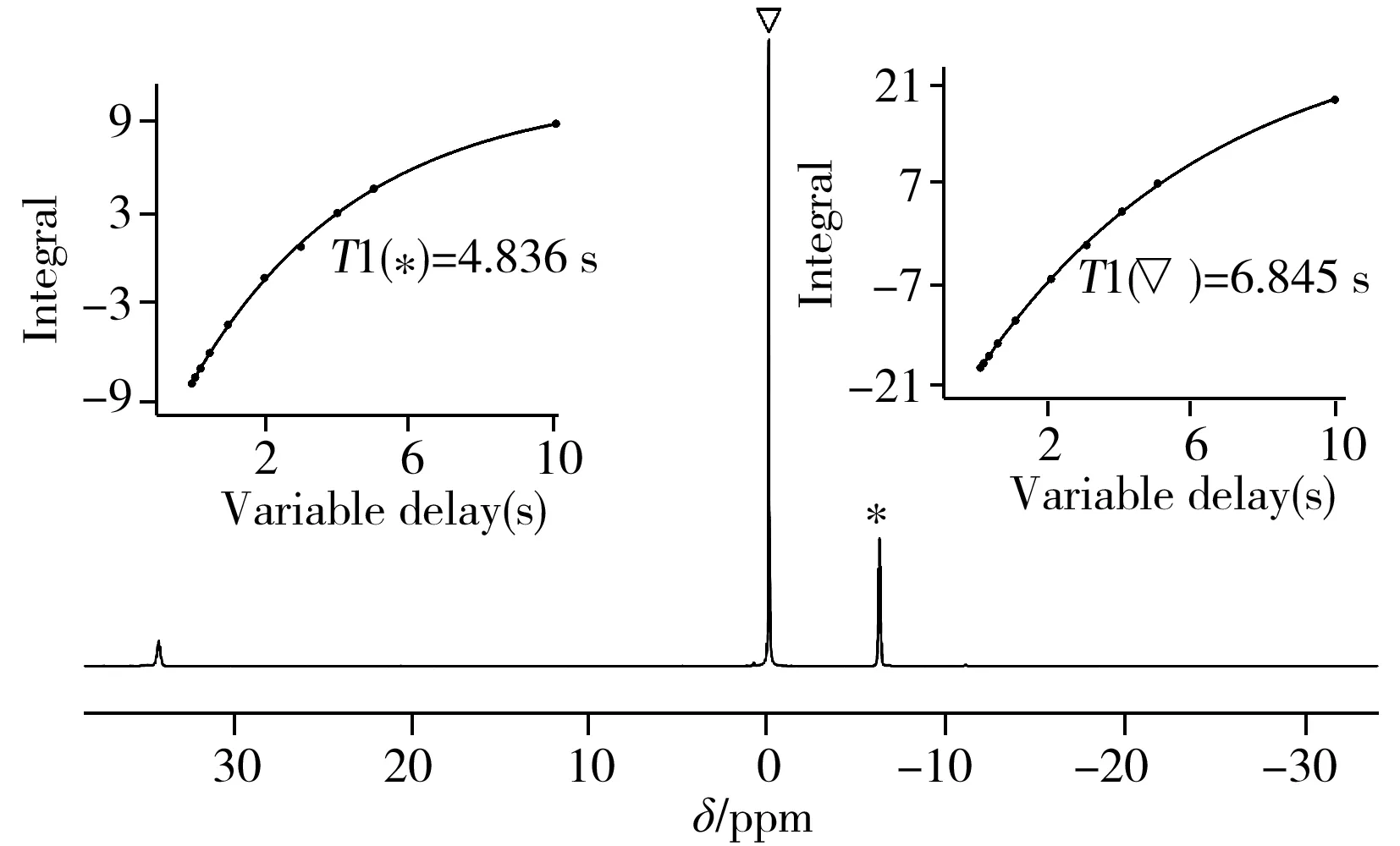
图6 TPPTS和KH2PO4在D2O-D2体系中的31P-NMRFig.6 31P-NMR spectra of TPPTS and KH2PO4 in D2O-D2insert:T1 fitting curve of the spin nuclear in TPPTS and KH2PO4(TPPTS和内标峰的纵向弛豫时间T1拟合曲线)
2.4.331P-NMR的定量分析31P的自旋量子数为1/2,自然丰度为100%,具有较好的信号响应。虽然31P的灵敏度不如1H,但31P-NMR的化学位移分布范围更宽,而且很多有机分子只含1种磷原子,所以谱峰重叠的概率很低,特别适合用作复杂体系中某种化合物的定量分析[18]。鉴于实际生产过程中TPPTS可能存在多种副产物,会导致氢谱谱峰严重重叠而无法定量,因此对TPPTS的31P-NMR定量方法进行了研究,并将其定量结果以与1H-NMR定量结果相互验证。
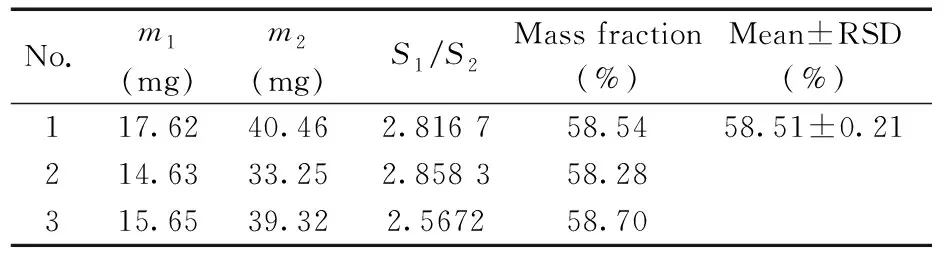
表3 TPPTS的31P-NMR定量分析结果Table 3 Quantitative analysis results of TPPTS using 31P-NMR
TPPTS的磷谱定量选取KH2PO4作为内标,图6为TPPTS和KH2PO4在D2O-D2体系中所采集的31P-NMR以及T1拟合曲线,其中把KH2PO4的化学位移δ定为0 ppm(▽标记),此时TPPTS的化学位移在δ=-6.2 ppm(*标记);经测定得到TPPTS磷原子的T1为4.836 s,内标磷原子的T1为6.845 s。为保证自旋体系的纵向磁化强度充分恢复,D1设置为35 s(尽管分析过程中采用30°脉冲进行激发)。另外,为了尽量减少偏共振效应,设置O1=-3.1 ppm。从表3中可看出3次测量测平均值为58.51%,相对标准偏差为0.21%,最终检测结果可表示为(58.51±0.21%)。测量结果偏差较小,且平均值与1H-NMR定量分析结果的平均值接近,仅相差0.21%,两种方法的相互验证说明TPPTS定量分析结果具有较高的准确性。
3 结 论
本文通过1H-NMR及31P-NMR技术建立了混合体系中TPPTS的有效定量分析。两种方法的结果接近(平均值相差0.21%),且各有优势:1H-NMR的定量分析耗时较短,而31P-NMR谱图的重叠概率小,解析简单,适合复杂混合体系中TPPTS的定量分析。本方法通过对文中定量分析过程以及一些关键参数进行设置,为其他化合物的定量分析提供了方法借鉴,对TPPTS的合成工艺以及Rh-TPPTS催化体系生产工艺的优化改进也具有重要指导意义。
猜你喜欢
云南化工(2023年7期)2023-08-01 07:59:34
现代仪器与医疗(2022年4期)2022-10-08 05:54:40
现代临床医学(2022年4期)2022-09-29 07:36:10
中国抗生素杂志(2022年7期)2022-08-18 03:22:36
大学化学(2021年2期)2021-04-09 11:15:32
保鲜与加工(2021年1期)2021-02-06 06:43:22
长春师范大学学报(2019年4期)2019-04-29 05:51:36
广东饲料(2016年5期)2016-12-01 03:43:22
百姓生活(2016年6期)2016-06-22 14:39:00
中国资源综合利用(2016年12期)2016-01-22 02:02:26
