
蛋清溶菌酶基因的密码子优化及其在毕赤酵母中的分泌表达
2020-08-17 07:55张赛南施柳佳廖志银王珍王首锋
浙江大学学报(理学版) 2020年4期
张赛南,施柳佳,廖志银,王珍,王首锋*
(1.浙江大学基础医学系,浙江杭州310058; 2.绿城农科检测技术有限公司,浙江杭州310052)
自1928 年弗莱明爵士发现青霉素以来,抗生素逐渐成为治疗各种细菌感染或致病微生物感染类疾病的“特效药”,大大提升了人类的健康生活质量[1]。近年来,由于抗生素的滥用加速了细菌的耐药性,由此衍生出DNA 污染、超级细菌等一系列问题[2]。溶菌酶(lysozyme,LYZ)最早由法国科学家尼克尔在枯草杆菌中发现,其可有效溶解细菌细胞壁的1-4 糖苷键,从而起到杀灭细菌的作用[3],是一种非特异性抗菌物质。市售溶菌酶类产品主要用于杀菌防腐[4],且无毒副作用,具有传统化学防腐剂不可比拟的优点,广泛用于各种食品[5]和饲料[6]中。
目前,市售的工业用蛋清溶菌酶(egg white lysozyme,EWL)主要通过化学方法从蛋清或蛋壳中提取,如盐析法、离子交换层析法[7]、超滤法[8]等。近年来,随着分子生物学和微生物表达技术的发展,通过微生物发酵大规模生产重组溶菌酶,可以极大地降低生产成本、提高产量。巴斯德毕赤酵母表达系统具有真核生物特有的生理体系,可对表达蛋白的加工、外分泌、翻译后修饰以及糖基化等进行调控,广泛应用于外源蛋白的表达[9]。多种类型的外源蛋白,如植物蛋白、动物蛋白在毕赤酵母中获得了成功表达,大大提高了生物蛋白的利用度。同时对毕赤酵母表达系统优化方法的相关报道也有不少[10-11]。
本文选用巴斯德毕赤酵母作为表达菌株,并通过蛋清溶菌酶密码子优化这一途径来获得高表达蛋清溶菌酶的重组酵母菌,对蛋清溶菌酶大规模工业化生产以及应用推广具有重要意义。
1 材料与方法
1.1 材 料
1.1.1 菌株和载体
藤黄微球菌(ATCC4698)购于美国Sigmam 公司,真核表达载体pPIC9K、毕赤酵母菌株GS115、大肠杆菌Top10、BL21 均为实验室保存。
1.1.2 试剂与工具酶
In-fusion clone 试剂盒(Clontech 公司生产)、高保真DNA聚合酶(PrimeSTAR HS DNAPolymerase)、限制性内切酶NotI、EcoRI、SnaBI、SalI 均购于宝生物工程(大连)有限公司;质粒小量抽提试剂盒、PCR 产物纯化试剂盒购于Axygen 公司;Bradford 蛋白定量试剂盒购于BIOMIGA 公司。Ampicillin、Kanamycin 和 G418 硫酸盐均购于生工生物工程(上海)股份有限公司;蛋清溶菌酶标准品和葡聚糖凝胶柱Sephadex G-50 购于美国Sigmam 公司;强酸性阳离子交换柱SPSepharose FF 购 于 Amersham公司。HD-3000 型电脑核酸蛋白检测仪购于上海嘉鹏科技有限公司。
1.1.3 菌株培养基
大肠杆菌-LB 培养基,酵母-YPD 培养基,发酵表达-BMMY、BMGY 培养基,具体的配制以及培养方法参见毕赤酵母多拷贝表达载体试剂盒说明书。
1.1.4 引 物
特异性引物:
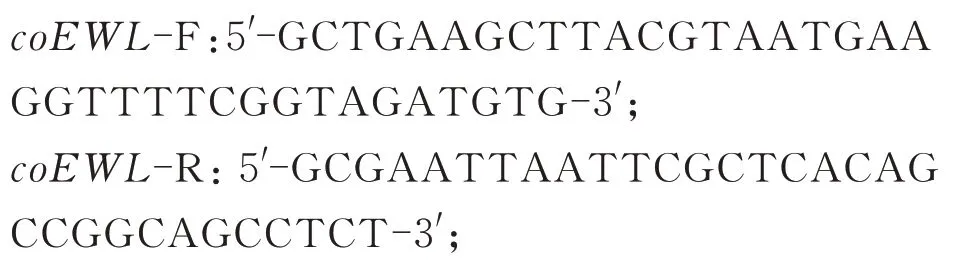
通用引物:
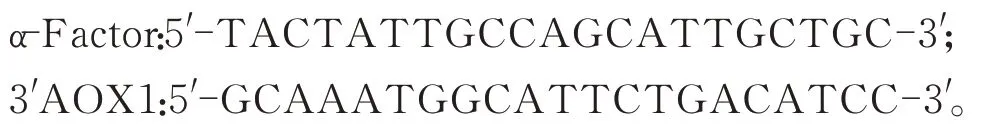
1.2 方 法
1.2.1 蛋清溶菌酶基因的密码子优化及重组表达载体的构建
从美国国家生物信息中心(NCBI)下载蛋清溶菌酶基因(Gene ID: 396218)的开放阅读框区段,在http://www.jcat.de/网站上选取酵母属作为表达菌株,替换该基因片段的个别密码子[12],目的基因序列的合成交由生工生物工程(上海)股份有限公司完成。通过设计与表达载体pPIC9K 具有同源区段的特异性引物,以coEWL为模板,在 95 ℃、3 min,95 ℃、30 s,68 ℃、10 s,72 ℃、30 s,30 个循环的 PCR反应条件下,高保真扩增出带同源臂的coEWL基因的ORF 区段。在In-fusion clone 酶作用下将其克隆到pPIC9K 中,构建重组表达载体pPIC9K-coEWL(见图1)。
在42℃条件下,将重组质粒热击转化至大肠杆菌BL21 感受态细胞中。用Kan+Amp 双抗性平板筛选阳性转化子。质粒抽提后进行双酶切验证,送至生工生物工程(上海)股份有限公司测序,检验其构建是否正确。具体操作参见毕赤酵母多拷贝表达载体试剂盒说明书。
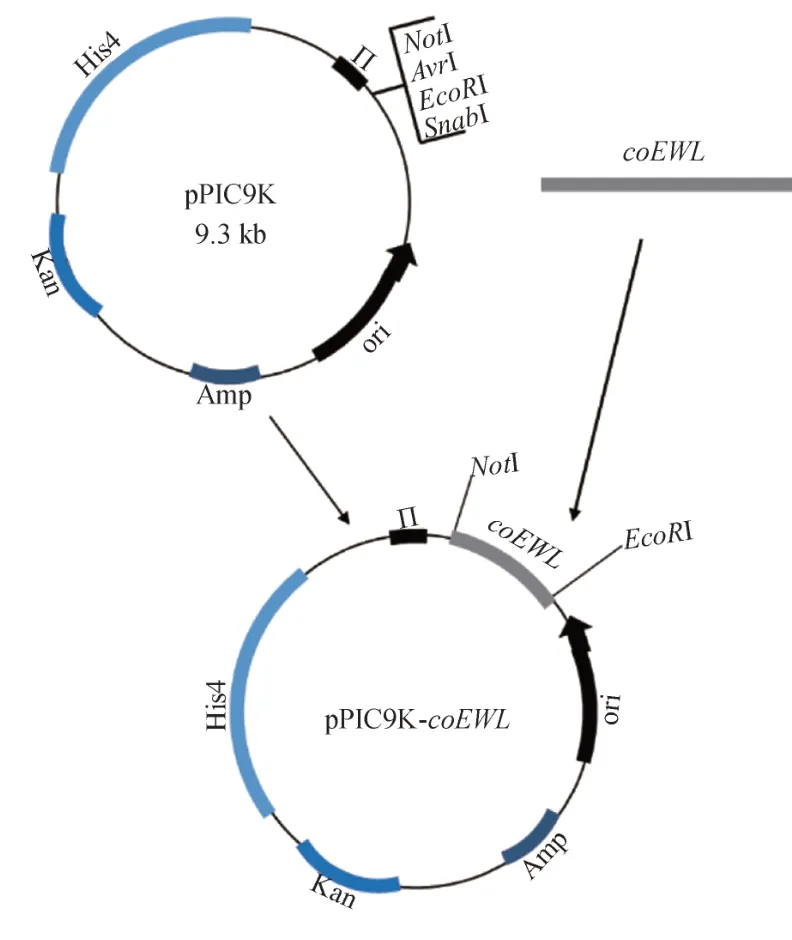
图1 重组表达载体pPIC9K-coEWL 的构建Fig.1 Construction of recombinant expression vector pPIC9K-coEWL
1.2.2 电转化、高拷贝酵母转化子的筛选及鉴定
选用SalI 酶切测序正确的重组表达载体pPIC9K-coEWL,获得线性质粒,利用同源重组将其整合至酵母基因组中。取OD600(酵母菌体在600 nm 处的光密度)为1.2 左右的酵母菌体20 mL,在8 mL混合溶液(10 mmol·L-1Tris-HCl, pH6.0,10 mmol·L-1LiAc, 0.6 mol·L-1山梨醇, 10 mmol·L-1DTT)中室温静置 30 min[13];4 ℃ 2 000 r·min-1离心,收集沉淀,用 1 mol·L-1山梨醇溶液清洗 3 次,最后添加 1 mol·L-1山梨醇溶液溶解 80 μL,制得酵母感受态细胞。取约10 ng 线性质粒与酵母感受态细胞混合,1.5 kV 电击后立即添加800 μL 山梨醇溶液,复苏1 h,涂布MD 平板,初步筛选His+转化子。
用无菌水冲洗MD 平板上生长的酵母菌落,将菌液稀释 104倍 ,并涂 布于梯度 G418(1~15 mg·mL-1)-YPD 平板上,选择高拷贝酵母转化子。随机选取长势良好的酵母转化子,放于单克隆YPD培养基中过夜培养,取200 μL 培养液离心,弃上清液,灭菌后用 ddH2O 清洗,沉淀 3 次,最后添加 50 μL无菌水混匀。取1 μL 上清作为菌落PCR 的模板,以α-Factor 和 3'AOX1序列作为引物 ,PCR 条 件 为95 ℃ 、5 min,95 ℃、30 s,56 ℃、30 s,72 ℃、2 min,30 个循环。用1%琼脂糖凝胶鉴定PCR 扩增产物。
1.2.3 重组酵母G-p-coEWL的诱导表达
挑选阳性酵母重组子(G-p-coEWL)单菌落,接种于 50 mL BMGY 培养基中,28 ℃,240 r·min-1培养18 h,离心后获得沉淀菌体。选取适量菌体,重悬于250 mL BMMY 培养基中,使菌液起始OD600在1.0 左右,相同条件下培养96 h。期间每隔12 h 取出5 mL 发酵液作为分析样品,立即离心弃菌体,保留上清液,于-20℃保存备用。同时向培养基添加适量体积无水甲醇,且维持甲醇浓度为0.5%。
1.2.4 表达蛋白的定性分析与酶活检测
取1 mL 上清液,用三氯乙酸(TCA)脱盐处理,然后进行SDS-PAGE 凝胶电泳。首先在样品中添加上清0.11 mL 的TCA 溶液,混匀于-20 ℃过夜,冷丙酮清洗沉淀,晾干加水溶解沉淀,添加上清缓冲液,98 ℃、5 min,即得电泳上样样品。配置15%分离胶以及5%浓缩胶,20A/板进行聚丙烯酰胺凝胶电泳,考马斯亮蓝染液染色,乙酸脱色液脱色,观察蛋白电泳结果。
Bradford 法测定发酵上清液中总蛋白浓度,配置牛血清蛋白(BSA)标准溶液,测定微球菌在595 nm 处的吸光度(A595)并绘制蛋白标准曲线,根据曲线计算发酵液蛋白量,详细操作步骤参见TaKaRa Bradford Protein Assay Kit 使用说明书。
采用比浊法[14]测定蛋清溶菌酶的酶活,以藤黄微球菌(ATCC4698)为指示菌,在溶菌酶的水解作用下,微球菌在450 nm 处的吸光度(A450)降低。一个溶菌酶的酶活单位被定义为在25 ℃、pH 6.2 条件下,使用藤黄微球菌悬浊液在450 nm 处引起吸光度每分钟变化量为0.001 所需的溶菌酶量。称取10~15 mg 藤黄微球菌溶于50 mL 无菌磷酸缓冲液(pH 6.2)中,倾斜混匀,放置在37 ℃培养箱中培养30 min。用PBS 溶液校对空白,起始底物溶液的A450控制在0.70±0.1 内。首先取蛋清溶菌酶标准品(Sigma,酶活 40 000 U·mg-1),用磷酸缓冲液(pH 6.2)配置浓度梯度为 1 000,500,250,125,50,25,0 U·mL-1的酶标准溶液,取 100 μL 酶标准溶液并与2.9 mL 菌悬液迅速混匀,记录A450:反应1 min 为A1,反应3 min 为A2,计算吸光度每分钟平均下降值以酶活为纵坐标,单位时间内吸光度变化量为横坐标,作蛋清溶菌酶酶活标准曲线。对发酵上清液做相同处理,根据酶活标准曲线计算酶活。
1.2.5 表达蛋白的纯化及其效果评价
为提升纯化效果,在37 ℃条件下,对发酵液上清做真空旋转浓缩处理。取浓缩样作为蛋白纯化上样样品。蛋清溶菌酶的等电点在11.0~11.5,在缓冲液pH 为7.4 时带正电荷,选用SP-Sepharose FF。蛋清溶菌酶的分子质量约为14.4 kDa,据此选用Sephadex G-50 进行蛋白纯化。同时用2 种方法对目的蛋白进行纯化,并比较2 种方法的效果。
离子交换层析。填料经HCl-NaOH 先后预处理后装柱,用 20 mmol·mL-1磷酸盐缓冲液(pH 为7.4)冲洗平衡,取 2 mL 浓缩样品,PBS 缓冲液稀释至10 mL,于4 ℃静置2 h,使目的蛋白充分带上正电荷。样品以1 mL·min-1的速度在压力泵上样,再转用缓冲液清洗(流速为2 mL·min-1)。电脑核酸蛋白检测仪实时检测并记录洗脱液的A280,洗脱至基线后,依次用含 0.1,0.2,0.3,0.4,0.5 mol·mL-1NaCl的磷酸缓冲液梯度洗脱,流速不变。根据洗脱峰分别收集洗脱液,于-20 ℃过夜,成固体后,放于真空冷冻干燥机中冻干36 h,收集粉末即得蛋清溶菌酶初步纯化产品。
分子筛层析。称取15 g 填料煮沸,过夜冷却后装柱,4 mL 浓缩样品上样,流速 0.2 mL·min-1,选用0.1 mmol·L-1醋酸铵溶液(pH 为 7.2)作为洗脱液,电脑核酸蛋白检测仪实时检测并记录A280,洗脱至基线。收集洗脱峰液体,于-20℃过夜,成固体后,真空干燥。取适量粉末溶解后进行SDS-PAGE 电泳,由条带初步判定纯化效果。
2 结果与分析
2.1 蛋清溶菌酶基因的密码子优化结果
蛋清溶菌酶基因的密码子与经优化后替换为碱基的基因比对,如图2 所示,同时添加了终止子TGA。图中,* 表示碱基一致,为半保守突变,空格表示保守突变。可见将蛋清溶菌酶基因替换为毕赤酵母所偏好的密码子后,更符合酵母自身的翻译体系,表达量可能会有所增加。
2.2 重组表达载体pPIC9K-coEWL 的构建
通过设计与毕赤酵母表达载体pPIC9K 具有同源区段的引物,以优化后的合成基因片段为模板,通过高保真PCR 扩增出带同源臂的coEWL基因的ORF 区段,获得422 bp 目的基因片段,其琼脂糖凝胶电泳结果如图3 所示。采用无缝克隆技术连接至酶切后pPIC9K 线性空载体,构建真核表达载体pPIC9K-coEWL。将重组质粒转至大肠杆菌BL21感受态细胞后,筛选出阳性转化子并抽提质粒,经BamHI、SalI 双酶切后,得到 2 614 bp 和 7 037 bp 目的基因片段,结果如图4 所示。测序结果显示重组质粒pPIC9K-coEWL构建正确。

图2 蛋清溶菌酶序列与优化后基因比对Fig.2 Alignment of egg white lysozyme sequence and optimized gene
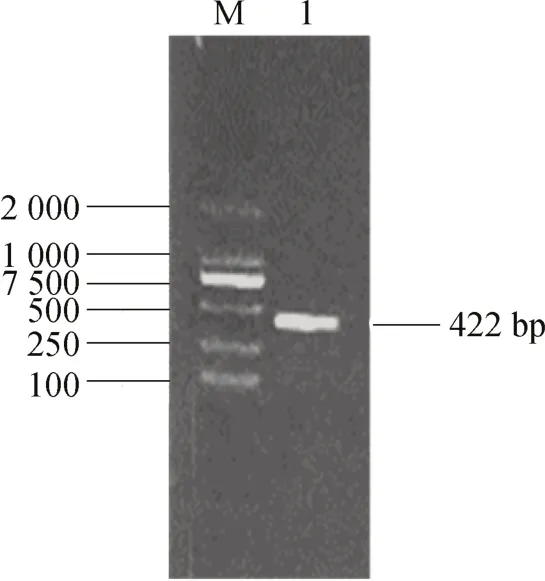
图3 coEWL PCR 扩增产物的琼脂糖凝胶电泳图Fig.3 PCR product of coEWL M:DL2 000 标志物,1:双酶切条带。
2.3 酵母转化子的筛选
电转化后的酵母菌液在MD 平板上初步筛选出His+转化子,如图5 所示,用无菌水清洗MD 平板上的菌落后,转涂G418 浓度梯度平板,30 ℃培养2~3d。鉴于酵母菌长势良好,逐步提升G418 浓度,考虑遗传霉素的高抗性可能与高拷贝酵母转化子的产生有关,最终在浓度为 15 mg·mL-1的 G418 YDP 平板上随机选取多个转化子,进行菌落PCR 验证。按上述方法制得模板,pPIC9K 通用引物扩增出的产物,电泳凝胶结果如图6 所示,其中2 200 bp 左右条带为 GS115 野生型的 AOX1 基因,890 bp 左右条带为目的基因片段(393 bp)与 pPIC9K 上 AOX1 基因(492 bp)之和,结果表明,携带有优化蛋清溶菌酶基因的线性化质粒已经重组到酵母基因组内,而且此菌株为Mut+型,可以有效利用甲醇进行发酵表达。将此菌株命名为G-p-coEWL,作为发酵表达菌株。
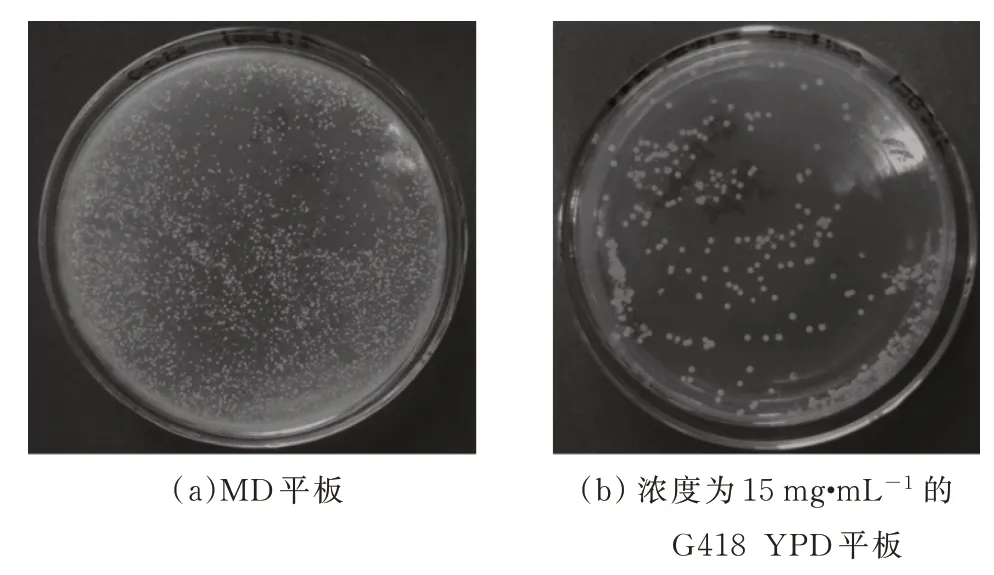
图5 毕赤酵母转化子平板鉴定Fig.5 Identification of Pichia pastoris transformant
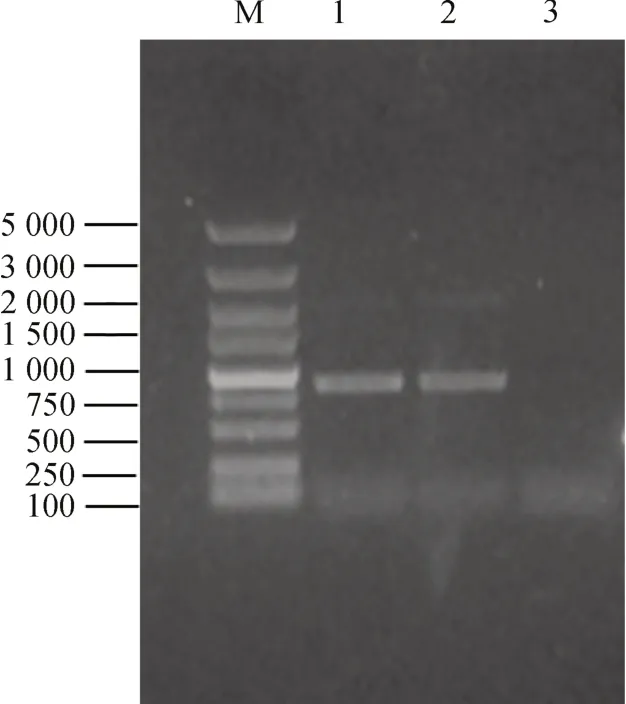
图6 毕赤酵母转化子菌落PCR 验证Fig.6 PCR product of Pichia pastoris transformant
2.4 重组菌株的诱导表达
G-p-coEWL首先利用BMGY 培养基培养足够量菌体,24~36 h 后取适量经离心后的菌体,重悬于250 mL BMMY 培 养基中 ,起始 OD600控制在 1.0 左右。诱导96 h,离心收集发酵上清液。经TCA 处理后,用SDS-PAGE 分析,可以观察到在14.4 kDa 处有逐渐变浓的条带,此为目的条带,如图7 所示。24 h 后蛋清溶菌酶开始表达,且表达量逐渐上升。
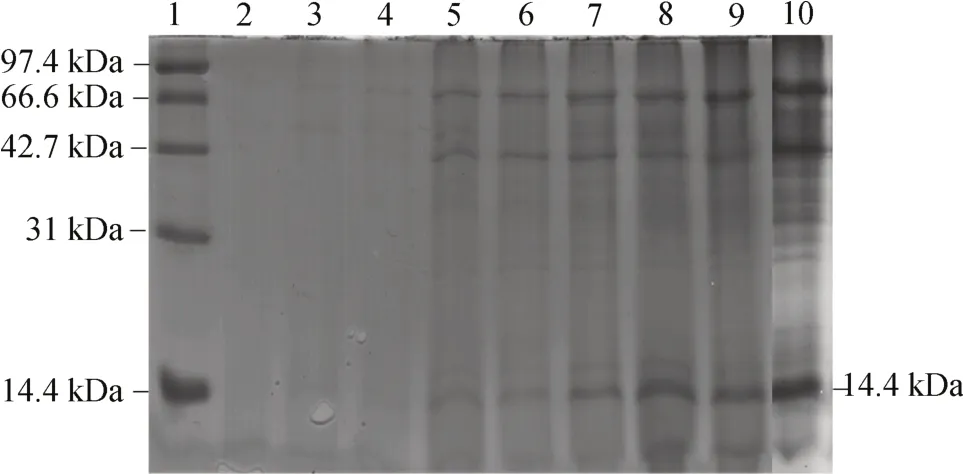
图7 发酵上清液蛋白的SDS-PAGE 分析Fig.7 SDS-PAGE analysis of fermentation supernatant protein
2.5 蛋清溶菌酶的酶活测定
按照上述试验方法绘制蛋清溶菌酶酶活标准曲线,测定取样样品酶活。以发酵时间为横坐标,每12 h 样品酶活为纵坐标作图,其结果如图8 所示。由图8 可知,在250 mL 摇瓶发酵表达中,酶活在36 h 处提升较明显,这可能与酵母自身系统的调控有关。72 h 后酶活趋于平稳,甚至有所降低。发酵上清液中酶活最高约为677 U·mL-1。
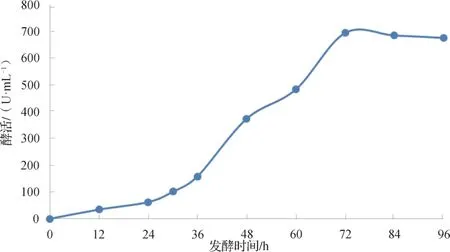
图8 发酵上清酶活变化Fig.8 Lysozyme activity of supernatant
2.6 蛋白的定量分析及纯化
首先,绘制BSA 标准曲线,然后,取96 h 发酵上清液20 μL 与考马斯染液反应,测得A595平均值为0.169,最后,根据BSA 标准曲线计算得到G-p-coEWL的发酵上清液中总蛋白浓度为0.607 mg·mL-1。
在阳离子柱纯化过程中,第一次上样后将未吸附蛋白洗脱液重复过柱,110 min 回归基线。换浓度梯度NaCl 缓冲液洗脱,每个浓度都在洗脱20 min 左右后出现洗脱峰,洗脱1 h 后换下一浓度溶液,最后高浓度清洗所有杂蛋白。从洗脱峰体积看,2 mL 样品中未吸附蛋白占比较大,为80%左右。这与填料体积以及最大吸附量有关。0.2 mol·L-1NaCl 溶液洗脱液中含有清晰的目的蛋白条带,同时含有其他杂蛋白条带。此pH 值下多种蛋白条带电荷相同。浓度为0.1 mol·L-1的洗脱峰面积大于浓度为0.2 mol·L-1的,说明洗脱峰体积与蛋白含量无直接关系。使用Sephadex G-50 纯化目的蛋白,共洗脱14.5 h。2 h 左右时出现第 1 正蛋白峰,5.25 h 时出现第2 叠加蛋白峰,此时吸光度还未降至零,11.13 h时出现面积较大的第3 峰,直到洗脱结束A280回到基线。
从纯化后蛋白的SDS-PAGE 分析图(见图9)可知,样品在过Sephadex G-50 柱子后,对目的蛋白的分离效果更佳,最终在14.4 kDa 处得到了单一的目的蛋白条带。而离子柱下经各浓度NaCl 溶液洗脱后的蛋白条带并不单一,没有达到很好的分离效果。
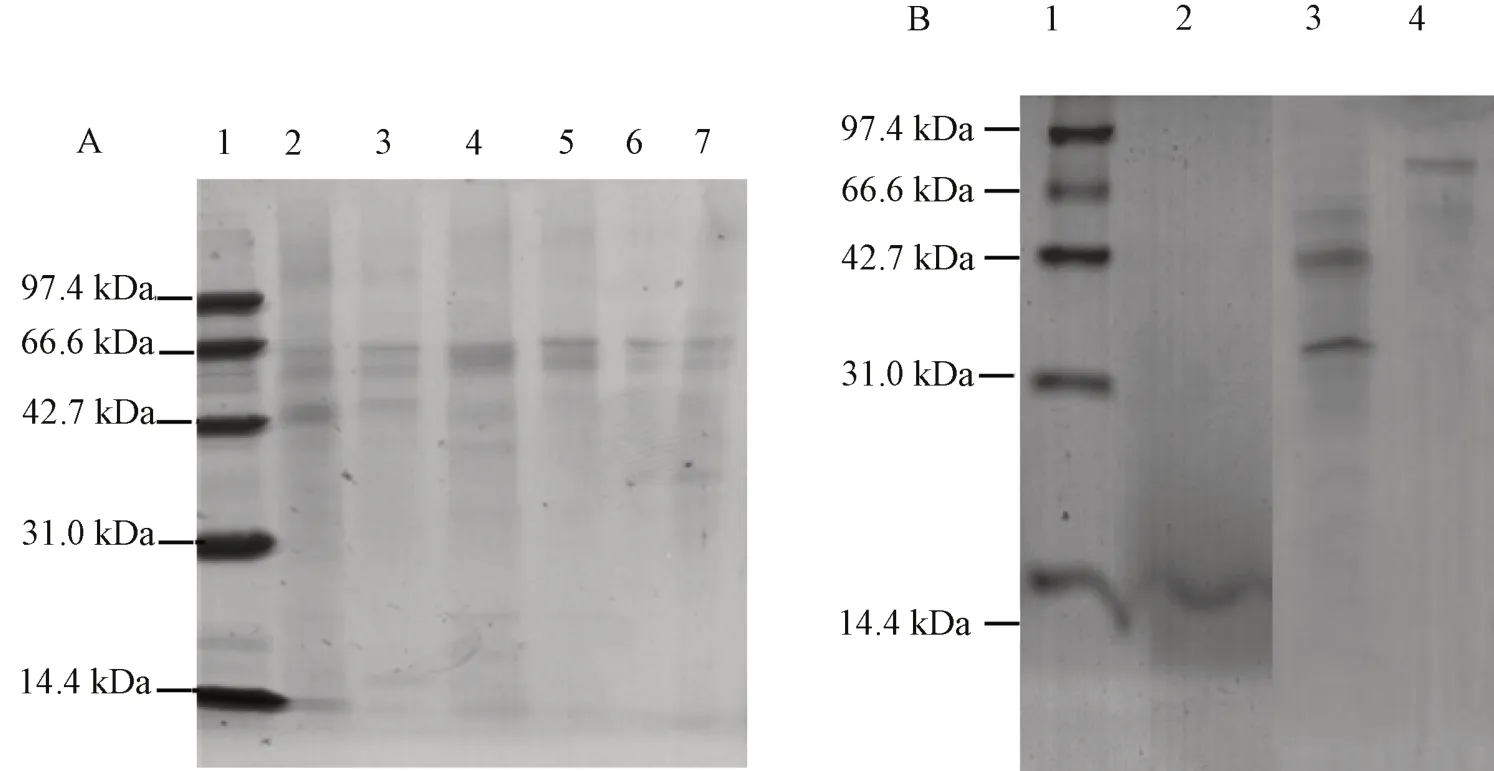
图9 纯化后蛋白的SDS-PAGE 分析Fig.9 SDS-PAGE analysis of purified protein
3 讨 论
巴斯德毕赤酵母作为较成熟的真核蛋白表达体系,已实现500 多种外源蛋白的表达,各种蛋白表达量暂不能完全满足市场需求[9]。研究人员针对此问题提出一系列解决对策,包括优化密码子、改造信号肽、添加强启动子等。本实验参照酵母菌属的密码子偏好性,通过改变外源目的基因的部分碱基序列(精氨酸序列不变),理论上可以提高在酵母细胞中的转录翻译频率,提高外源蛋白的表达量。YANG等[15]将黑曲霉的2 型脂肪酶基因理性优化后,在毕赤酵母中获得了191 U·mL-1的表达量,在原16.5 U·mL-1的水平上提升了近10 倍。本实验首先对蛋清溶菌酶的密码子进行优化,替换了106 个碱基,氨基酸序列未改变,旨在提高外源基因在酵母细胞中的转录效率。
在制备酵母感受态细胞过程中,相比于原实验方法中只使用山梨醇溶液浸泡清洗酵母菌体,本实验首先采用混合处理液对酵母细胞进行浸泡,再经山梨醇溶液清洗获得酵母感受态细胞,其他电转化条件不变。原方法几乎没有筛选出转化子。改进实验方法后,最终得到了一株可抗高浓度G418(15 mg·mL-1)的转化子G-p-coEWL。在现有可见报道中,电转化获得酵母重组子可抗G418 的浓度最高为5 mg·mL-1[16]。毕赤酵母高拷贝表达试剂盒手册中提到,可抗浓度为4 mg·mL-1G418 的酵母转化子体内拷贝数达7~12 个,高抗性的产生可能与外源基因插入的拷贝数有关[17]。在G-p-coEWL中需要插入的外源基因拷贝数有待通过RT-PCR 等实验进一步探究。
重组酵母菌G-p-coEWL,经摇瓶发酵虽能成功表达溶菌酶蛋白,但产量和活性没有达到理想的发酵水平,可能与发酵方式和发酵参数未优化有关,以此为基础,后期通过优化发酵参数(特别是通气条件)或采用发酵罐高密度发酵,预计发酵水平可以提高50~100 倍。张洋[18]将海参i-型溶菌酶及基因优化后转入毕赤酵母进行发酵表达,在摇瓶条件下只获得了8.79 U·mL-1的表达产物,采用5 L 发酵罐表达后,酶活提高了近52 倍。另外,G-p-coEWL菌株为Mut+型,能高效利用甲醇进行外分泌表达,发酵周期比Mut-型菌株缩短近一半,目的蛋白外分泌表达也使后期发酵产品纯化更简单,这些优势极大地降低了溶菌酶的生产成本,对溶菌酶大规模工业化生产具有至关重要的意义。
同时,本文比较了Sephadex G-50 和SPSepharose FF 这两种纯化方法,可以看出样品在过Sephadex G-50 柱后,对目的蛋白的分离效果更佳,但是Sephadex G-50 柱花费时间较长,所以仍需寻找合适的蛋白分离方法。
猜你喜欢
学与玩(2022年10期)2022-11-23
畜牧兽医学报(2022年4期)2022-04-24
广州化工(2022年20期)2022-01-01
齐鲁工业大学学报(2021年1期)2021-12-30
福建农业学报(2021年6期)2021-08-18
百姓生活(2021年4期)2021-05-06
消费导刊(2020年35期)2021-01-28
教学考试(高考生物)(2020年4期)2020-11-18
发明与创新·中学生(2019年6期)2019-06-26
安徽农业科学(2018年1期)2018-05-14
