
伪狂犬病病毒变异株US3基因失活株的构建与生物学特性分析
2020-08-12 09:52吕家轩张传健侯继波费荣梅王继春
畜牧与兽医 2020年8期
吕家轩,张传健,侯继波,费荣梅,王继春
(1. 南京农业大学动物医学院,江苏 南京 210095;2. 江苏省农业科学院动物免疫工程研究所,江苏 南京 210014;3. 江苏省农业科学院国家兽用生物制品工程技术研究中心,江苏 南京 210014;
伪狂犬病是由伪狂犬病病毒(pseudorabies virus,PRV)感染猪、羊、牛等家畜以及多种野生动物而引起的一种传染病[1-2],临床症状包括发热、瘙痒、脑脊髓炎和呼吸及神经系统障碍等[3]。PRV对仔猪危害极大,对15日龄内仔猪致死率可达100%。2000年之后,我国通过基因缺失疫苗与生物检测技术已对PRV取得了良好的控制。但2011年以来,一种新型PRV变异株在全国多地的猪场中暴发流行,其毒力与传播力显著高于传统毒株[4-5],甚至有感染人的报道出现[6],这不仅对猪场的传染病防控提出严峻考验,也对公共卫生安全形成严重的威胁。
疫苗是控制疫病的最有效的方式,研究发现,传统Bartha-K61株对变异株不能提供完全的保护[4]。应用同源病毒研制的弱毒活疫苗(缺失gE/gI、gE/TK或者TK/gE/gI)能够有效抵抗变异株攻击[7-9],但是其安全性还不够高[8]。这暗示一些其他基因可调控PRV的毒力。PRVUS3基因属于复制非必须基因,是病毒的重要毒力基因,编码产物是丝/苏氨酸激酶[10]。研究表明PRVUS3能够降解Bcl2相关转录因子1(Bcl2 associated transcription factor 1,Bclaf1)进而抑制Ⅰ型IFN反应,减弱IFN-α抑制病毒复制的作用[11]。单纯疱疹病毒1型的US3基因可下调感染细胞表面主要组织相容性复合物Ⅰ(major histocompatibility complex,MHC-Ⅰ)的表达,阻滞其向CD8+T细胞递呈抗原,进而抑制细胞免疫[12]。PRVUS3基因依靠其激酶活性,参与下调PRV感染猪睾丸(swine testis,ST)细胞表面MHC-Ⅰ的表达[13]。目前,变异株US3基因的失活对其毒力的影响还未见系统的报道。
本研究应用细菌人工染色体技术(bacterial artificial chromosome,BAC)构建了PRV变异株AH02LAUS3基因失活株PRV-US3mut,并对其生物学活性进行了分析。研究发现,通过点突变起始密码子构建的PRV-US3mut在ST细胞中复制稳定。US3基因可能介导PRV感染ST细胞的早期复制,可抑制PRV感染ST细胞早期MHC-Ⅰ的mRNA转录,US3基因失活可降低PRV对小鼠的致病性。本研究为PRV变异株的基因缺失新靶点提供了重要依据。
1 材料与方法
1.1 毒株、菌株、细胞和试验动物
PRV AH02LA株由江苏省农业科学院动物免疫工程研究所从安徽六安某猪场的PR发病猪分离、鉴定并保存[14];含BACPRV-G的大肠杆菌GS1783由本实验室以PRV AH02LA为亲本毒株制备并保存[15];ST细胞由本实验室保存,生长于DMEM培养基(含10%胎牛血清+1%青链霉素),37 ℃,5% CO2培养。BALB/c小鼠购于南京市江宁区青龙山动物繁殖场(合格证编号:NO.201929986)。
1.2 主要试剂
LATaq DNA聚合酶、高保真DNA聚合酶、DNA Marker、琼脂糖凝胶DNA回收试剂盒、反转录试剂盒和荧光定量试剂盒购自TaKaRa公司;新生牛血清、胰酶和DMEM购自Gibco公司;脂质体转染试剂盒购自Thermo Fisher公司。
1.3 引物的设计与合成
以下引物由本实验室设计并由擎科生物科技(南京)有限公司合成(表1)。

表1 引物序列
1.4 重组菌株BACPRV-G-US3mut的构建
根据文献[8]所述,应用引物US3mut-Kan F/R,以卡那霉素(Kan)抗性基因为模板,扩增包含US3点突变起始密码子与Kan抗性基因序列的PCR片段。PCR片段经回收和Dpn I酶切后电转入含BACPRV-G的感受态细胞GS1783中进行第一轮同源重组,再经第二轮重组,将KAN抗性基因敲除,获得重组BAC,命名BACPRV-G-US3mut。应用引物US3 F/R对US3基因起始密码子突变进行PCR和测序验证。
1.5 重组毒株PRV-US3mut和亲本恢复毒株PRVrec的获得与鉴定
使用通用型柱式基因组DNA提取试剂盒,提取PRV AH02LA病毒基因组DNA,应用引物H89 F/R PCR扩增两端带有同源臂的gE/gI基因序列。碱裂解法提取BACPRV-G-US3mut的DNA。通过脂质体转染试剂盒将约3 μg带有同源臂的gE/gI基因分别与2μg BACPRV-G-US3mut和BACPRV-G的DNA共转染单层ST细胞。在转染后1~2 d观察到在紫外光(488nm)激发下不发出荧光的病毒蚀斑,经3轮挑斑筛选,获得纯化的病毒,命名为PRV-US3mut和PRVrec。通过PCR、基因测序鉴定gE/gI基因序列是否正确。
1.6 PRV-US3mut遗传稳定性测定
重组毒株PRV-US3mut在ST细胞上连续传至15代,用通用型柱式基因组DNA提取试剂盒提取F15代病毒基因组DNA,应用US3 F/R PCR扩增US3基因序列,通过测序鉴定US3基因起始密码子突变是否稳定。
1.7 PRV-US3mut体外生长特性测定
将重组毒株PRV-US3mut、亲本毒株PRV AH02LA和亲本恢复毒株PRVrec分别以感染复数(multiplicity of infection,MOI)为0.001接种长满单层的ST细胞上,分别于接种后6、12、24、36、48和72 h收集培养物上清和细胞混合物,经-70 ℃和37℃冻融3次后,12 000 r/min离心10 min取上清,检测病毒的TCID50,共重复3次。使用软件Graphpad Prism 5绘制生长曲线,通过软件SPSS 11.0进行统计学分析。
1.8 病毒感染ST细胞早期MHC-I转录水平检测
将PRV-US3mut和PRV AH02LA按MOI稀释度为10接种于单层的ST细胞,置于5% CO2恒温培养箱中37℃孵育1 h后,吸去上清液,PBS洗涤3次,换成细胞维持液(含3%胎牛血清的DMEM培养基)进行培养。接种后6 h收集细胞,使用Trizol试剂提取细胞RNA,采用Nanodrop 2000对RNA的浓度和纯度进行检测,OD260/OD280应介于1.9和2.1之间。随后,采用1%甲醛变性凝胶电泳对RNA完整性进行检测,选取RNA质量较高的样品,进行下一步试验。使用PrimerScript TM RT Reagent Kit(TaKaRa,Japan)进行cDNA合成。反应体系包括反转录酶、寡核苷酸、RNA、引物、缓冲液和灭菌双蒸水。反应步骤为37 ℃水浴15 min,85 ℃反应15 s,获得cDNA置于-20 ℃保存。通过荧光定量试剂盒对β-actin和MHC-Ⅰ基因进行qRT-PCR扩增。反应体系为:2×SYBR Green PCR Master Mix 10 μL,上游引物和下游引物各0.4 μL,cDNA 2.0 μL,ROX Reference Dye 0.4 μL和灭菌双蒸水6.8 μL。反应条件为:95 ℃ 30 s;95 ℃ 5 s,60 ℃ 30 s,共40个循环;95 ℃ 15 s,60 ℃ 1 min,95 ℃ 15 s。共重复3次。采用2-ΔΔCt法分析基因表达并使用软件SPSS 11.0分析显著性差异。
1.9 PRV-US3mut对小鼠致病力测定
试验选取65只BALB/c小鼠,分组如表2所示。取滴度为106TCID50/mL的PRV AH02LA、PRV-US3mut和PRVrec3种毒株,倍比稀释至10-5,取10-2~10-5进行攻毒。攻毒组每只小鼠皮下接种0.2 mL病毒稀释液,对照组接种等量PBS溶液。攻毒后统计死亡情况,连续观察14 d。按Reed-Muench法计算PRV AH02LA、PRV-US3mut和PRVrec对小鼠的LD50。

表2 小鼠试验分组
2 结果
2.1 BACPRV-G-US3mut的获得与鉴定
通过第一轮重组,将BACPRV-G的US3基因起始密码子进行点突变,同时插入KAN抗性基因作为筛选标记。经过氯霉素、卡那霉素抗性筛选后,得到重组BAC(BACPRV-G-US3mut-Kan)。核酸电泳结果(图1)显示,以BACPRV-G-US3mut-Kan为模板扩增的US3基因序列相较于BACPRV-G的US3基因序列,条带大小增加约1 000 bp。然后进行第2轮重组,敲除Kan基因,PCR(图1)和测序表明US3起始密码子点突变成功。
2.2 重组毒株PRV-US3mut和亲本恢复毒株PRVrec的获得与鉴定
将带有同源臂的gE/gI基因DNA片段,分别与BACPRV-G-US3mut和BACPRV-G的DNA共转染ST细胞,在拯救病毒的同时,将BAC中的mini-F基因替换为变异株AH02LA株的gE/gI基因序列。转染后1 d观察到在紫外光(488 nm)激发下不发出荧光的病毒蚀斑,经3轮挑斑,获得2株纯化的病毒,分别命名为PRV-US3mut(图2)与PRVrec(图3)。PRV-US3mut与PRVrec的gE/gI基因经PCR(图4)和测序验证,与变异株AH02LA一致。

M. DL 5000 DNA Marker;1. US3;2. 插入KAN基因;3. 敲除KAN基因图1 KAN抗性基因的插入与敲除鉴定
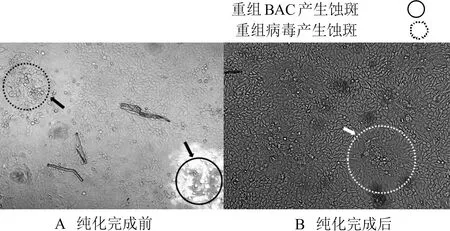
图2 重组毒株PRV-US3mut纯化

图3 亲本恢复毒株PRVrec纯化

M. DL 8000 DNA Marker;1. gE/gI;2. PRV-US3mut;3. PRVrec图4 重组病毒PRV-US3mut与PRVrec gE/gI基因序列PCR鉴定
2.3 PRV-US3mut遗传稳定性测定
将PRV-US3mut在ST细胞中连续传至15代,对F15代PRV-US3mut的US3基因进行PCR和测序验证,结果显示PRV-US3mut起始密码子仍保持点突变状态。
2.4 PRV-US3mut、PRV AH02LA和PRVrec体外生长特性分析
将重组毒株PRV-US3mut、亲本毒株PRV AH02LA和亲本恢复毒株PRVrec分别以MOI为0.001接种单层ST细胞,在指定时间点收集上清与细胞混合物检测TCID50。生长曲线如图5所示,相对于亲本毒株和亲本恢复毒株,PRV-US3mut感染ST细胞12 h无病变发生,暗示US3基因可能参与病毒增殖的早期阶段。在接种后36~48 h,3种毒株的滴度都达到峰值,PRV-US3mut的最高滴度(107.17TCID50/mL)相较于PRV AH02LA(107.63TCID50/mL,P=0.085)和PRVrec(107.37TCID50/mL,P=0.410)略有下降,但无显著性差异。

图5 PRV-US3mut、PRV AH02LA和PRVrec体外生长特性
2.5 PRV-US3mut对ST细胞MHC-Ⅰ转录水平的影响
PRV-US3mut和PRV AH02LA感染ST细胞6 h通过qRT-PCR检测,MHC-Ⅰ的mRNA转录水平。如图6所示,PRV-US3mut组的MHC-Ⅰ mRNA转录水平显著高于AH02LA组(P=0.020),但显著低于正常细胞组,表明US3基因能够抑制感染细胞的MHCⅠ转录,且有其他蛋白共同参与此过程。
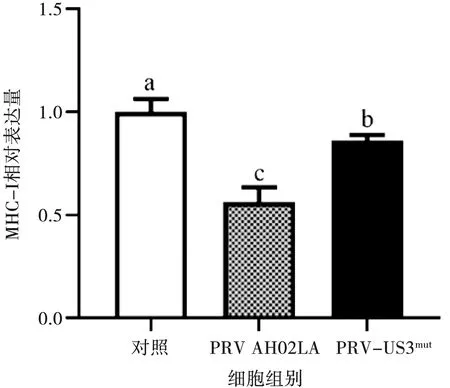
注:图中字母不同表示差异显著(P<0.05)图6 PRV-US3mut与PRV AH02LA感染ST细胞6 hMHC-Ⅰ的mRNA转录水平
2.6 PRV-US3mut、PRV AH02LA和PRVrec对小鼠致病力分析
如表3所示,亲本毒株PRV AH02LA与亲本恢复毒株PRVrec对小鼠的LD50分别为10-3.26/0.2 mL和10-3/0.2 mL。PRV-US3mut攻毒组小鼠全部存活,表明US3基因可能介导PRV对小鼠的致病性。
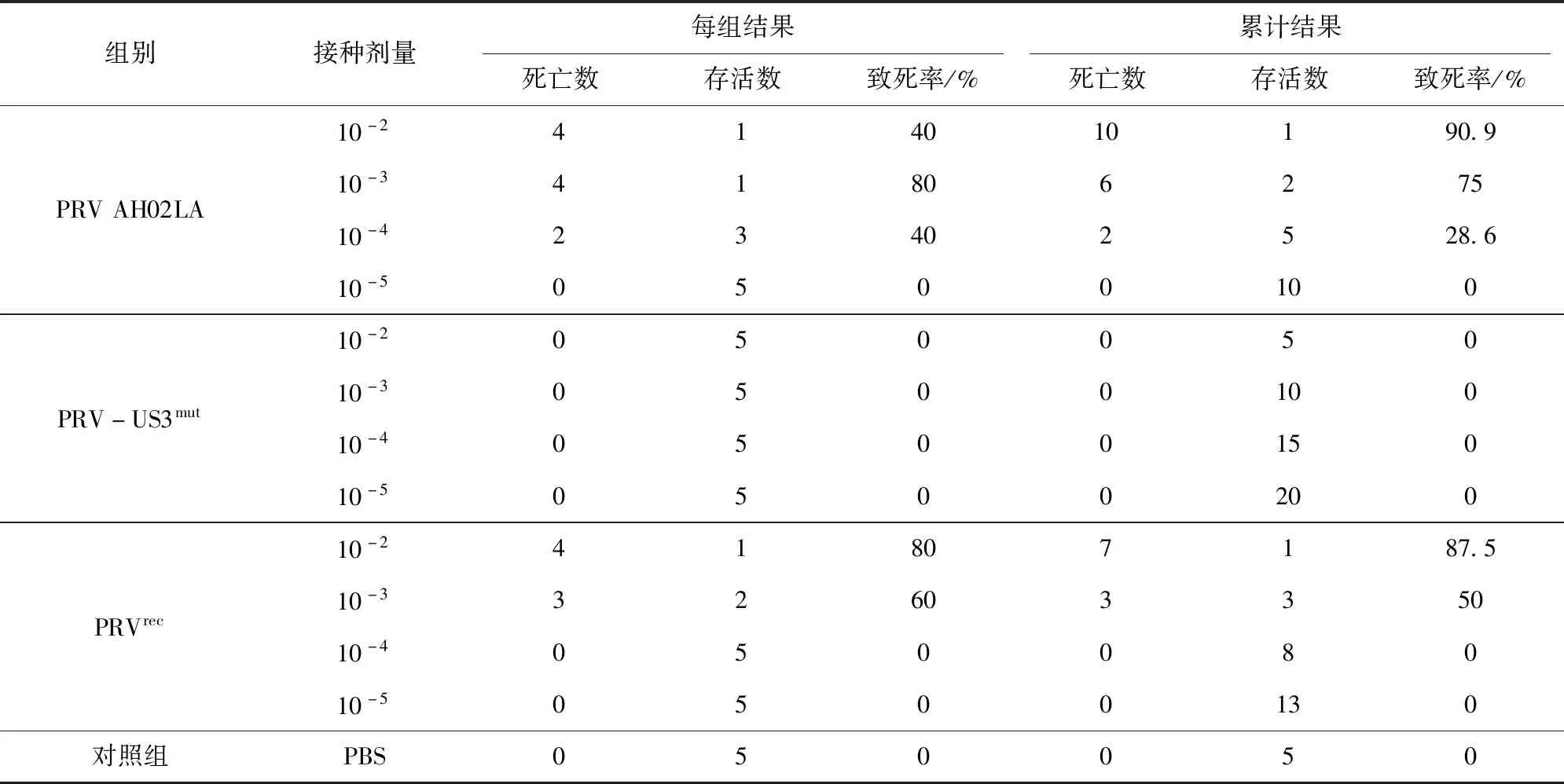
表3 小鼠死亡情况结果
3 讨论
2011年以来,一种PRV变异株在我国暴发流行,变异株不仅造成仔猪发病死亡,母猪流产,同时对生长猪和育肥猪致病力较强,甚至引起死亡,Bartha-K61株不能提供完全的保护力[4],应用同源病毒研制的弱毒活疫苗(缺失gE/gI、gE/TK或者TK/gE/gI)能对变异株提供更好的保护[7-9],但是现有变异株活疫苗的安全性还不够高,暗示存在其他基因影响PRV的毒力。PRVUS3基因属于复制非必须基因,是重要的毒力基因。目前,变异株US3基因的失活对其毒力的影响还未见报道。本研究应用基因工程技术将US3基因失活,探究其是否与PRV变异株毒力具有相关性,可配合原有的基因缺失弱毒疫苗进一步降低PRV毒力。
PRV吸附并侵入靶细胞后,合成病毒成分并装配成成熟的病毒粒子,最后释放并感染邻近细胞[10]。本研究中,PRV-US3mut的生长曲线相较于亲本毒株PRV AH02LA,毒力呈迟发型,提示US3基因可能介导PRV体外增殖的早期阶段。PRV与细胞质膜外表面的相应受体结合后,释放携带部分内层间质蛋白的核衣壳进入胞浆,此过程中US3基因可能对核衣壳的转运起促进作用。核衣壳以出芽方式从细胞核内膜排出至核周腔,UL34基因能够介导初级核衣壳与细胞核膜的融合[16],而后核衣壳又在US3基因的作用下进入细胞质,US3基因还可增强UL34基因的活性[17]。有研究表明,PRVUS3基因缺失株的初级病毒粒子聚集在细胞核内膜的内凹处[18]。此外,US3基因诱导细胞骨架重排和细胞延伸,从而促进PRV向周围细胞扩散[19]。基于前人和本研究结果可推断:US3基因失活影响PRV感染细胞后核衣壳向细胞核的转运以及核衣壳向细胞质外排,进而延缓病毒粒子组装,同时抑制PRV向周围细胞扩散。
对小鼠致病力研究发现,PRV-US3mut组小鼠全部存活。先前研究表明,PRVUS3基因具有多种毒力作用。US3基因可调节细胞凋亡内外途径相关蛋白的活性,进而抑制细胞凋亡[20]。US3基因是PRV中发现的唯一能够编码具有抗凋亡活性的基因。此外,US3基因可引起自然杀伤细胞表达的CD300a与靶细胞表面的磷脂酰丝氨酸结合,从而抑制自然杀伤细胞的免疫功能[21]。另有研究发现,US3基因通过降解Bcl-2来抑制Ⅰ型IFN反应[11]。US3基因可下调PRV感染ST细胞表面MHC-Ⅰ基因的表达量[13],本研究也证实,US3基因对PRV变异株感染ST细胞的MHC-Ⅰ基因转录存在抑制作用。为精确US3基因是否失活,今后的研究需通过Western blot进行验证。综上所述,US3基因对非特异性免疫与特异性免疫均有抑制作用,这在一定程度上解释了US3基因失活减弱了PRV对小鼠的致病性,为PRV基因工程活疫苗的开发提供了新的参考位点。
综上,本研究应用基因工程技术构建了PRV变异株US3基因失活株PRV-US3mut。生长动力学试验发现PRV-US3mut在感染ST细胞早期(6和12 h)无病变发生,在感染ST细胞24 h之后,与亲本毒株PRV AH02LA生长动力学相似,表明US3基因可能参与病毒增殖的早期阶段。对小鼠致病性试验发现US3基因失活显著减弱PRV对小鼠的致病性。本研究为PRV变异株的弱毒株疫苗的研制奠定了基础。
猜你喜欢
云南化工(2021年6期)2021-12-21
长江蔬菜(2021年12期)2021-04-04
农药科学与管理(2019年6期)2019-11-23
中国果业信息(2019年11期)2019-01-05
西南农业学报(2016年5期)2016-05-17
华南农业大学学报(2015年5期)2015-12-04
河南科技(2015年2期)2015-02-27
新疆农垦科技(2014年10期)2014-02-28
现代检验医学杂志(2014年5期)2014-02-02
食品科学(2013年19期)2013-03-11
