
小胶质细胞表型和功能的多样性与阿尔茨海默症
2020-08-12 09:00国佳莹石京山
遵义医科大学学报 2020年3期
国佳莹,石京山
(遵义医科大学 基础药理教育部重点实验室暨特色民族药国际合作联合实验室,贵州 遵义 563099)
在中枢神经系统(central nervous system,CNS)中,胶质细胞可分为小胶质细胞(Microglia,MG)、星形胶质细胞(Astrocyte,AS)和少突胶质细胞。MG是CNS的天然的免疫效应细胞,在生理情况下,MG处于M0静息状态,并发挥免疫监视、免疫防御和修复组织的作用[1]。当脑损伤或受到炎性刺激时,MG处于M1、M2两种表型的动态平衡中,但大脑受到过度刺激后,MG转化成M1表型并且调控多种炎性介质的释放,如:肿瘤坏死因子-α(Tumor necrosis factor,TNF-α)、白介素-1β(IL-1β)和一氧化氮(NO)等,这些介质的不断产生和积累从而造成了周围神经元的损伤[2]。同时,受损的神经元释放多种毒性物质,如β-淀粉样蛋白(Aβ)和α-突触核蛋白(α-synuclein)等,然而这些毒性物质能够进一步诱导MG形成M1表型并且又再次释放神经毒性物质。所以,受损的神经元和M1表型的MG之间构成了一种恶性循环,尤其是对神经退行性疾病,例如在阿尔茨海默病(Alzheimer’s disease,AD)的发生发挥重要作用[3]。受到外界过度刺激的MG转化为M1表型后,M1表型MG能够调控炎性因子的释放,并且形成级联放大反应从而导致神经系统疾病的发生,体内一些活性物质可以调控M0向M1表型的转化或者促进其向M2表型转化[4];M2表型的MG通常被称为“守卫保护大脑者”能够及时清除脑有害物质。M2表型能够发挥吞噬作用、调控抗炎因子的表达增加比如:促进抗炎因子IL-10等表达,从而修复损伤的神经元[5]。
1 MG的表型分型
在健康的脑组织中,MG是一种极具可塑性和多向性的细胞类型。他们以不同的形态存在,即“静息型MG”(M0)和“活化型MG”,后者又分为M1型和M2型两种活化细胞,维系着CNS微环境的平衡[6]。在CNS内环境发生改变时,如炎症、损伤、抗原入侵等,M0型MG转变为激活的M1和/或M2型MG,以保护CNS免受神经损伤或致病性入侵和随后的神经炎症反应[7-8]。进一步研究发现,MG的M1型又称为“经典激活”,主要发挥促炎作用,M1表型的生物因子标志主要为TNF-α、IL-1β、IL-6、IL-12和CD16/32;M2型又称为“替代激活”,主要发挥抗炎和修复作用,而M2表型的生物因子标志主要是IL-10、Arg-1和CD206[4]。
2 MG的表型转化
当CNS受到刺激时,M0型MG迅速被激活,转化为免疫原性表型M1和抗炎症表型M2型MG。研究表明,M1型MG能够释放神经毒性物质,诱发炎症反应;M2型MG具有多种免疫保护功能,在脑中起到免疫消除作用,并能促进中枢神经系统组织的修复和再生[9]。根据基因表达谱,M2型MG又被细分为3种不同的激活状态即:M2a、M2b、M2c。M2a型MG是通过选择性激活产生,并通过抗炎因子的分泌和碎片的吞噬作用促进免疫消除,通过产生神经营养因子如胰岛素样生长因子-1(IGF-1)促进组织再生;M2b型MG被免疫复合物介导的Fcγ受体刺激激活,其特征是IL-12分泌下调,而IL-10分泌增多;M2c型MG是M1型MG在糖皮质激素或IL-10作用下失活而成,其分泌特征主要由转化生长因子(TGF-β)和鞘磷脂激酶的表达所主导,发挥促进组织修复和基质重构[10-11]。最近的研究发现,静息和活化状态的MG功能转化是由相关的信号传导和生长因子进行调控:在静息状态下,MG通过激活周围神经元和AS分泌的几个关键细胞因子信号轴,如TGF-β、小细胞浸润因子和集落刺激因子1(CSF-1)受体来维持细胞环境的稳态。当脂多糖(LPS)或干扰素-γ(IFN-γ)刺激时,会促进MG的M1型形成,分泌炎性介质:TNF-α和IL-1β等,从而对健康脑细胞造成损伤[12]。然而,在白介素-4(IL-4)作用下,却可以促进M2表型形成,主要引起抗炎反应,释放白介素-10(IL-10)等介质,促进轴突再生,发挥神经保护作用[13]。研究发现,在体外细胞培养中,免疫荧光标记的MG的M1和M2表型特征性标志物,随时间增加,不同表型所占比值也在发生动态变化。具体来说,在维持大脑神经稳态的平衡过程中,初始M2型比M1型表现强,其对伤口愈合和抗炎修复发挥重要作用;但在慢性炎症后期,M1型占主导地位,导致神经损伤[14]。因此,在维持CNS稳态的过程中,调节小胶质细胞M1/M2表型的转化具有重要意义(见图1)。
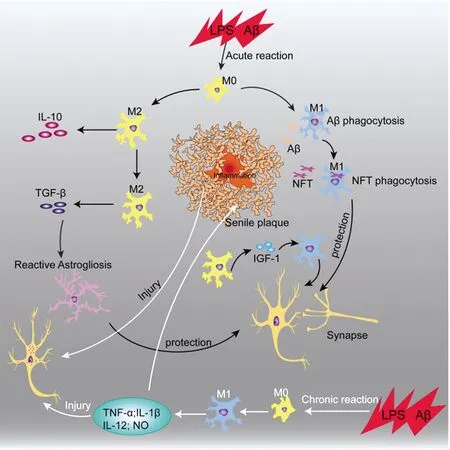
图1 MG表型转化及功能示意图
3 MG在CNS微环境调节作用
MG在CNS微环境中是重要的调节因子。当遇到外界刺激时,MG的高度动态和反应性分泌使其能对细胞外界信号做出快速反应,即MG通过中和病原体或者一些致病性肽如Aβ和分泌抗炎因子如IL-10或生长因子,来修复和防止大脑的过度损伤,在急性炎症期间可以对大脑起到免疫保护作用[15]。静止状态下的MG通过吞噬作用,清除病原体和细胞碎屑来调节CNS的稳态,而过度炎症会导致神经细胞损伤和神经退行性疾病的发生。现在研究发现,不同阶段的MG反应能够干预和改变炎症并在神经系统退行性疾病防治中开辟新的方向[16]。实际上,在生理条件下,急性炎症反应之后,MG信号通路会产生一个负反馈循环,从而有效地消除炎症。在脑损伤模型中,MG首先上调IL-6等炎症细胞因子的表达,并刺激IL-10和TGF-β的表达快速升高,通常在小鼠损伤脑组织7天后达到峰值[17]。而IL-10和TGF-β是重要的细胞因子,能介导向周边的MG由M1向M2表型转化,以重建中枢神经系统稳态微环境。TGF-β还能促进神经营养因子的产生,并刺激早期AS增生,以减少寡聚体Aβ突触毒性。此外,MG和AS的TGF-β 信号通路已被证明可以减少海马神经元中AD相关的树突棘丢失,从而并提高记忆能力[18]。
4 MG与AD
AD是痴呆症的主要病因。由于老龄化人口的急剧增加,它在发达国家的发病率不断上升,这是对现代医学最昂贵的挑战之一[19]。面对这一挑战,对AD的发病机制有更全面的了解已成为现在关注的热点。最近的研究显示,越来越多的证据表明,作为大脑固有的免疫系统细胞的MG发挥重要作用,MG对病原体和炎症的反应现在已明确。在寻找AD基因修饰因子的过程中,全基因组关联研究发现,大部分AD的遗传风险存在于MG而非神经元中表达的基因,如APOE、TREM2、CD33、BIN1、INPP5D、PICALM等一系列致病基因[20]。根据越来越多的文献报道,MG在AD中对个体细胞因子和趋化因子的有保护和损害作用,说明其具备“双刃剑”的功能;研究也指出MG的神经营养因子和神经源性因素在CNS中所扮演的不同角色,所以MG在AD的发病机制中如何发挥作用是目前许多学者的关注点[21-22]。研究发现,Aβ沉积、Tau蛋白过度磷酸化、补体系统的激活等都能够激活MG从而促使AD发生。当Aβ沉积形成斑块后通过激活MG受体或当Aβ部分片段与C1q补体蛋白受体相结合形成攻膜复合体(membrane attack complex,MAC)促进神经炎症并导致AD的发生[23-24]。此外,MG内丝裂原激活蛋白激酶(MAPK)途径被激活并释放大量炎症因子,同时Tau蛋白发生过度磷酸化发生异常聚集,形成神经纤维缠结(neurofibrillary tangles,NFTs)促进AD发生[25]。
4.1 MG与神经炎症 在AD微环境中,MG反馈系统的缺失可导致其促炎和抗炎作用的平衡偏离生理平衡。新发现的证据表明,在这种慢性神经炎症环境,先天免疫通路被激活,抑制MG的表达和神经毒性因子的增加,导致了神经元的损失[26]。利用基于MRI方法的神经图像和胶质反应、细胞因子改变、神经发生的组织学进行评估,研究者发现,在TgAPP/PS1小鼠中,MG和AS在6个月大时被激活并聚集在Aβ斑块周围,在6~12个月大时随年龄增长显著增加。聚集的MG显著上调促炎因子的产生,包括TNF-α、iNOS和IL-1β[27]。研究发现,在APP/PS1模型小鼠中,核转录因子(NF-κB)炎症通路被激活,TNF-α、IL-1β、IL-6等炎症因子表达含量明显增加,这些炎症介质又能够进一步激活NF-κB信号通路,从而促进炎症联级放大反应[28]。此外,MAPK信号通路被激活后,p38-MAPK能够诱导小胶质细胞释放大量炎症因子、粘附因子等,同时也促进MG本身凋亡[29-30]。采用p38抑制剂阻断炎症通路的发生能够减缓AD进展。另外,当MG被激活后,过氧化物酶体增殖物激活受体(PPAR)信号通路也会被激活并能够抑制炎症作用,促进MG由M1型向M2型转化,并分泌IL-4和IL-10等抗炎因子以及神经营养因子从而抑制炎性反应的发生[31]。当PPARδ与NF-κB亚基结合,并抑制NF-κB蛋白转录过程,从而减少炎症介质的释放,最终发挥防治AD的作用[32]。Toll样受体4(TLR4)的抑制已被证明与免疫反应和脑损伤有关。文献报道与C57BL/6野生型小鼠相比,APP/PS1转基因AD小鼠TLR4水平显著升高,同时MG的炎性因子表达显著升高。TLR4 抑制剂 TAK-242可显著改善神经功能,降低Bax水平,显著降低M1标记物iNOS和TNF-α水平,而M2表型标记物TREM2和Arg-1在体内外表达增加。与此同时,在Aβ25-35所致的AD模型中,炎症标志物MyD88、NF-κB-p65和NLRP3的升高,NLRP3升高可被NLRP3-siRNA或TAKC-242预处理所抑制。这些结果表明,抑制TLR4具有神经保护作用,并促进了AD中MG从炎性M1表型向保护性M2表型的转变[33-34]。
4.2 MG与补体系统激活 在各种神经系统疾病中,补体通路过度激活可导致神经元损伤。新近研究发现,在淀粉样变异蛋白(PS2APP)和具有病变的Tau蛋白(Tauopathy)Taup301小鼠模型中,MG中典型补体成分和中心成分C3表达增加。通过去除C3阻断补体功能,可以减少PS2APP小鼠斑块相关的突触丢失,改善Taup301小鼠的神经元丢失和脑萎缩,改善神经生理和行为。此外,在AD患者的大脑中发现,神经元突触中C3蛋白升高,并且脑脊液中C3蛋白的水平也增加,并与Tau蛋白有一定的联系,此外,过度刺激C3补体受体后,使得MG,尤其是M1型MG激活,并释放大量炎症因子,破坏微环境的平衡从而促进AD发生[35]。这些结果表明,在大脑损伤和病变的环境中,激活的补体系统可以通过Tau蛋白病理引起的神经退行性变。虽然AD的特征是β-淀粉样蛋白斑块(Aβ)和Tau蛋白缠结,但现在对补体的研究主要集中在淀粉样变模型上[36]。聚集的Aβ也可以激活补体,通过MG吞噬作用导致突触被枝剪甚至丢失。值得注意的是,全身系炎症可激活大脑中的MG的TLR4、NLRP3炎症小体和补体,从而导致CNS神经炎症、Aβ聚集、突触丢失和神经退行性变[37]。根据现在机制研究发现,在β-淀粉样蛋白斑块(Aβ)的AD模型小鼠中,其大脑神经元的突触出现丢失,并在Aβ斑块沉积的周围出现MG大量激活,随后补体系统激活,C1q等补体蛋白参与神经炎症反应发生[38]。MG表面高表达C1q补体受体蛋白,Aβ前体蛋白(APP)经β-和γ-裂解酶分解而成的Aβ1-16可与C1q受体结合,其结合的亲和力高,对补体系统激活作用明显[39]。当大量的Aβ与MG表面的C1q受体结合后,补体系统被过度激活形成MAC,并对正常的组织造成损坏作用,使得细胞凋亡和溶解;同时,MAC也能够激活M1型MG,并产生大量的炎症因子,形成链式级联放大反应的恶性循环,从而导致AD发生[40]。研究发现,将人工合成的Aβ42多肽疫苗注射进入APP/PS1模型小鼠后,通过检测发现小鼠血清中具有高浓度的抗Aβ42的抗体,且脑内Aβ沉积含量显著降低从而降低了AD的发生[41]。在补体受体系统中,当MG表面的C3补体受体激活后,可以调节MG在初期进行吞噬细胞分泌毒性物质、清除Aβ等,进而维持神经元微环境平衡[42]。
4.3 MG与Tau蛋白磷酸化 大量的Aβ沉积在神经组织周围形成斑块和神经细胞的Tau蛋白过度磷酸化和异常聚集形成神经纤维缠结(Neurofibrillary tangles,NFTs)是AD主要病理特征[43-44]。在正常的神经组织微环境中,Tau蛋白是微管相关蛋白(Microtubule associated proteins,MAPs)组成成员之一,微管蛋白与正常的Tau蛋白相结合可以促进微管的形成、稳定和延长从而发挥微管在神经组织中的蛋白物质运输功能并维持正常的神经细胞骨架结构[45]。但在AD患者脑中或者APP/PS1小鼠中,正常的Tau蛋白发生磷酸化从微管骨架上解离下来,导致过度磷酸化Tau蛋白发生异常聚集,从而形成NFTs刺激MG释放大量的炎症因子导致AD的发生。研究发现,采用野生型的Syn基因蛋白与Tau蛋白竞争性地结合微管组装蛋白,从而能够有效控制Tau蛋白发生过度磷酸化并影响微管的功能作用发挥[46]。Tau蛋白过度磷酸化在MG内激活mTOR(雷帕霉素靶蛋白)蛋白受体或者Toll样受体,并通过GSK-3β、MAPK等炎症信号通路进一步促进AD发生。利用雷帕霉素可减轻Tau蛋白过度磷酸化和异常聚集[47-48]。同时在激活MG表面的Toll样受体和IL-1受体后,通过p38-MAPK途径可以引起炎症因子大量释放,Tau蛋白发生过度磷酸化进行病理化扩散并引起神经元的损伤和记忆空间受损从而促进AD发生[49]。有研究报道可以通过研发Tau蛋白疫苗来减少磷酸化Tau蛋白的聚集形成和对神经元损害作用;同时,采用Toll样受体抑制剂能够促进MG由M1型向M2型转化,并释放抗炎因子,减少磷酸化Tau蛋白的形成和异常聚集,从而延缓AD发生[50]。
4.4 MG与吞噬作用 髓样细胞触发受体(TREM2)在CNS中仅在MG表面表达。研究表明,激活TREM2对Aβ沉积和磷酸化Tau蛋白的吞噬清除作用具有一定调控作用[51]。在AD小鼠中,体外给予IL-4可激活TREM2,导致溶酶体和微管相关蛋白1轻链3(LC3)II/I表达升高,说明MG自噬水平升高,由此显著下调半胱天蛋白酶募集域含蛋白9(CARD9)和TLR4的表达水平,通过削弱CARD9-TLR4通路,抑制TLR4介导的促炎作用,这一研究结果提示,TREM2可能是治疗AD神经炎症的潜在药物靶点[52]。进一步研究发现沉默TREM2基因,AD患者或在APP/PS1小鼠模型中的Aβ沉积越多,且MG被激活,以及释放大量炎症因子促进AD发生[53]。增加TREM2受体表达,发现MG周围的Aβ沉积明显减少,且MG对Aβ的吞噬作用增强,释放的抗炎因子IL-10以及神经营养因子等明显增加,提示TREM2基因可以促进MG由M1型向M2型转化[54]。此外,研究发现,在APP/PS1模型小鼠中,TREM2基因缺陷或转导受损时使磷酸化Tau蛋白表达含量明显增多,并会发生异常聚集形成NTFs,MG的吞噬功能下降[55]。因此,通过过表达TREM2,能够促进MG由M1型向M2型转化,进而增加MG吞噬磷酸化Tau蛋白功能[56]。
5 展望
目前的一系列神经系统疾病的发病机制和治疗手段并未完全清楚,此篇综述探讨的是:在正常的生理条件下,MG处于M1、M2两种表型的动态平衡中,但受到外界刺激后,M1表型的MG通过激活补体和释放炎症因子等导致疾病发生;而M2表型的MG发挥吞噬沉积的Aβ、磷酸化的Tau蛋白作用来抑制疾病的发生。因此,通过调节MG的M1、M2表型转化功能状态来防治疾病的发生,但是目前对于药物和治疗方案来调控M1、M2表型转化的报道较少,所以可以以此篇文章,为治疗神经系统疾病的药物研发和MG表型转化抑制疾病作进一步的探讨。
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
河北果树(2021年4期)2021-12-02
世界科学技术-中医药现代化(2021年5期)2021-11-05
世界科学技术-中医药现代化(2021年12期)2021-04-19
昆明医科大学学报(2021年1期)2021-02-07
天津医科大学学报(2021年1期)2021-01-26
中华养生保健(2020年7期)2020-11-16
医药前沿(2020年20期)2020-11-10
河北农业科学(2019年6期)2019-03-21
