
STK25的生物学特性及其与肿瘤关系的研究进展
2020-08-11 06:55:48张亦超吴英珂王卫星
中国普通外科杂志 2020年7期
张亦超,吴英珂,王卫星
(武汉大学人民医院 普通外科,湖北 武汉 430060)
哺乳动物绝育20(sterile twenty,Ste20)样激酶家族是典型的丝裂原活化蛋白激酶大家族,可通过经典的MAPK信号通路调节细胞生长、分化、死亡、应激、炎症反应等多种重要的细胞生理/病理过程[1-2]。该家族共有60个成员,在哺乳动物中已有超过30个成员被研究和记录,基于序列同源性,这些成员又被划分为p21活化蛋白激酶(p21-activated protein kinases,PAK)和生发中心激酶(germinal center kinases,GCK) 2个小家族,而STK25(又称作YSK1或SOK1)、MST3(mammalian Ste20-like kinase 3)、MST4共同构成GCK家族第III亚族(GCKIII)[3-5]。在研究细胞极性改变及凋亡时,学者发现GCKII亚族(MST1、MST2)与GCKIII亚族存在极高的相似性,因此这两个亚族在影响疾病的发生与发展的研究中常被共同讨论[6]。
近年来,肿瘤细胞的营养与代谢治疗得到了广泛关注,通过营养干预影响肿瘤的代谢过程,主要以调节碳水化合物、蛋白质/氨基酸及脂肪三大宏量营养素摄入为主,包括减少葡萄糖的供给,提高蛋白质供给,选择合适的脂肪酸及生酮饮食,最终抑制肿瘤细胞的生长,甚至诱导其死亡[7-8]。研究[9-11]表明,GCKIII与多种人类疾病的发生发展密切相关,而STK25不仅可以影响正常细胞的糖脂代谢,通过激活下游各类信号通路直接/间接诱导细胞的增殖/死亡,在肿瘤细胞中的表达水平与肿瘤的生长和增殖也呈现一定的相关性。本文将分别从STK25及家族部分成员的结构特点、生物学作用及肿瘤学效应等方面进行综述,探讨STK25与肿瘤发生发展关系的研究进展,为最终将其应用于肿瘤的临床治疗提供借鉴。
1 STK25的结构特点
GCKIII编码基因是由12个外显子组成,在体内多种细胞内均可广泛表达,如睾丸、胃肠道、肝脏等。STK25是该家族第一个被克隆的成员,研究过程中发现STK25编码基因位于染色体2q37.3,其mRNA编码的426-残基蛋白则来自外显子2-12,外显子1则编码了5’端的非翻译区域[5,12]。GCKII、III亚族的一级结构均包含一个保守的蛋白激酶催化结构域和一个可变的调控结构域,分别位于N末端和C末端。N端的激酶催化结构域又划分为11个子域,具有丝/苏氨酸蛋白激酶的全部特征。而由于GCKIII C端调控结构域的限制导致该亚家族成员在Ste20样激酶家族中长度最短[5]。另外2个亚族功能区还包含了核定位序列(nuclear localization signal,NLS)、核输出序列(nuclear export sequences,NES)、caspase裂解位点及部分未命名的同源序列(图1)[13],决定了该蛋白在细胞中的分布、从细胞质到细胞核的转移、核易位等改变。凋亡过程中,caspase酶可通过作用于caspase裂解位点,使NLS序列高度暴露,实现激酶的核转移[14-15]。进一步研究显示GCKIIIC端调控域与PDCD10(programmed cell death 10/cerebral cavernous malformation 3,CCM3)的N端同源二聚体高度相似[16],并且已在心血管疾病及脑海绵状畸形疾病中发现MST4的C端(346-416)残基与PDCD10共结晶,提示该亚族可能也与心血管疾病、脑畸形、细胞存活或凋亡相关[17]。
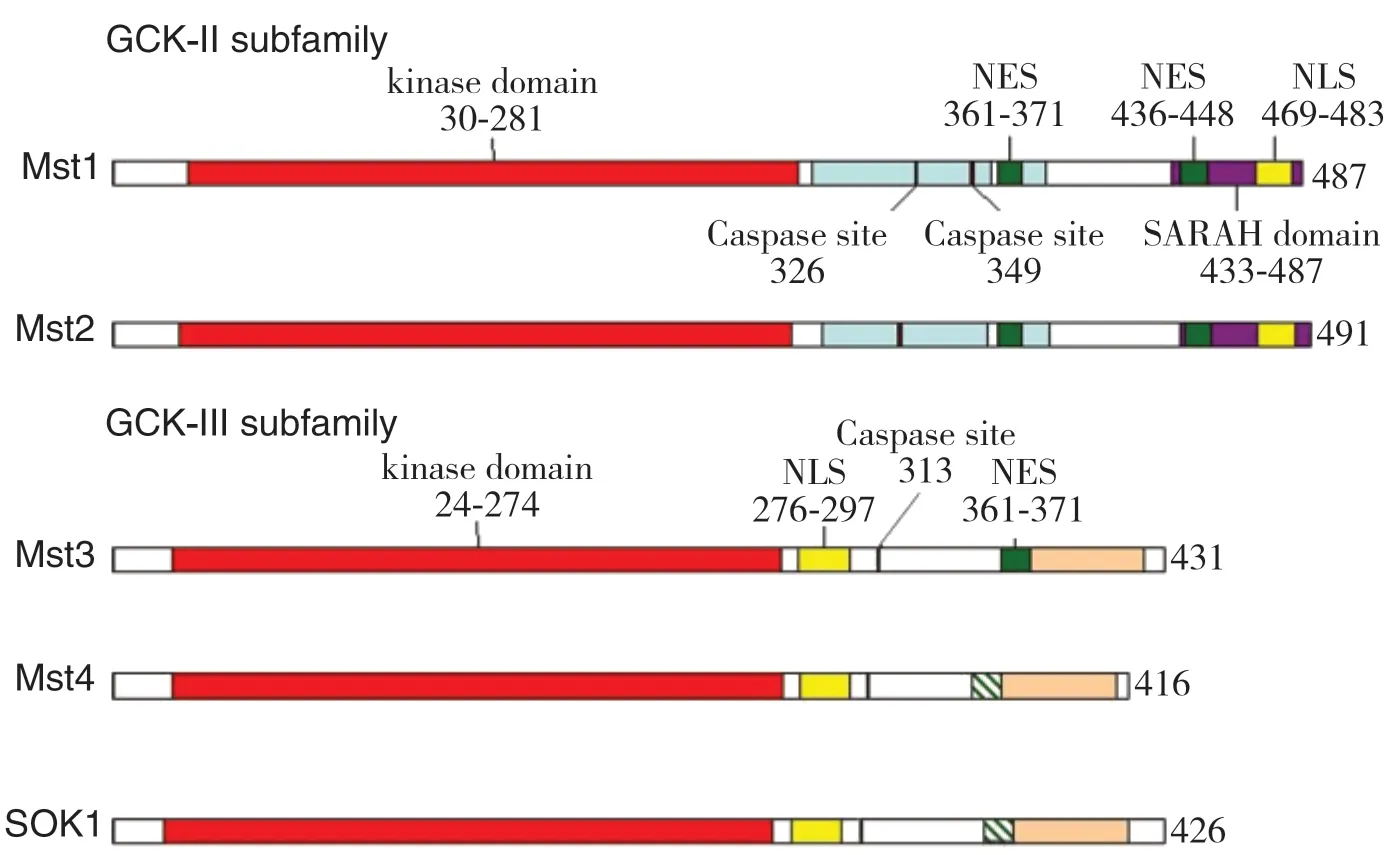
图1 哺乳动物GCKII和GCKIII蛋白的结构Figure1 Structure of the mammalian GCKII and GCKIII proteins
2 STK25调节细胞增殖/死亡/迁移
STK25在小鼠、大鼠和人类组织器官中均有广泛表达,正常状态下,STK25定位于细胞的高尔基体中,以细胞迁移所需的信号级联方式工作,即高尔基体蛋白GM130靶向结合STK25的C端(270-302),通过磷酸化下游特异性底物14-3-3ζ后,直接调节细胞极性及迁移[18]。而在受到外界刺激(ROS、化学性缺氧)时,STK25首先自磷酸化使自身被切割,产生一个35 kDa的N端片段,该片段可从高尔基体转移到细胞核中诱导高尔基体解体,同时磷酸化下游特异性底物14-3-3ζ的Ser58位点,释放14-3-3ζ/ASK1(apoptosis signal-regulating kinase 1)复合物中的ASK1,激活JNK和p38-MAPK,最终导致细胞死亡[19-20]。另外,GCKIIIC端调控域与PDCD10在结构上高度相似,而PDCD10在细胞迁移、增殖、凋亡和高尔基体组装中的研究已较为成熟[21-23],因此STK25与PDCD10之间是否有相互作用值得关注。研 究[24-26]发现PDCD10在体外可直接与STK25的C端(333-426)及FAS相关磷酸酶1(Fas associated phosphatase-1,FAP-1)连接,其结合域使PDCD10去磷酸化,从而负调节PDCD10活性和下游信号传导,可能诱导Fas介导的细胞凋亡。同时在伤口愈合实验中也发现,PDCD10正常状态下可与STK25、GM130形成相互作用形成复合物,维持高尔基体稳定及细胞功能,而PDCD10缺失的细胞不能正确地重新定向高尔基体和中心体,并显示出细胞迁移受损,该过程也表现出GM130/STK25复合物稳定性被破坏及底物14-3-3ζ去磷酸化,不难推断PDCD10对高尔基体的影响至少部分是通过STK25介导的14-3-3ζ磷酸化所产生[22]。因此改变STK25的活性或表达水平,通过STK25/GM130/PDCD10调节细胞增殖/凋亡/极性可能为肿瘤治疗提供新方向。
3 STK25调节能量代谢
机体主要通过以三大营养物质“糖类、蛋白质、脂肪”为基础的能量代谢维持自身正常的生理功能,而能量代谢的异常和紊乱与疾病产生密切相关。由于生活水平的提高,糖脂代谢异常导致的疾病也逐年递增。在寻找糖尿病治疗靶点时发现,STK25可作为异位脂质沉积、全身葡萄糖和胰岛素稳态的中央调节器,而非脂肪组织中的异位脂质沉积可推动胰岛素抵抗效应,最终可诱导2型糖尿病的发生与发展[9,27]。研究显示,2型糖尿病患者骨骼肌中STK25的表达水平较正常人显著上升,而抑制STK25的表达可增强胰岛素敏感性,提高肌肉细胞对葡萄糖的摄取[28]。此外,在两项动物学研究中也发现与野生型幼鼠相比,过度表达STK25的转基因小鼠表现出高胰岛素血症、全身葡萄糖和胰岛素稳态受损[29],而STK25基因敲除小鼠可抵制高脂饮食诱导的葡萄糖不耐受和胰岛素抵抗效应[30],但具体的分子机制尚不清楚。
在非酒精性脂肪肝炎/肝病(non-alcoholic steatohepatitis/Non-alcoholic fatty liver disease,NASH/NAFLD)的研究中发现,STK25是影响该病进展及预后的关键因子。研究人员在以胆碱不足的饮食饲养小鼠,模拟NASH中肝细胞损伤、脂肪变性、纤维化等病理改变时发现STK25敲除后的小鼠该病理改变不显著,甚至可阻断该病理进程,而且不同时期NASH患者肝穿刺检测STK25 mRNA表现一致性[27]。进一步研究证实,对照组小鼠肝和人肝细胞中的STK25表达增加,并且其可通过抑制脂解活性抑制β-氧化和甘油三酯外排,促进肝内脂滴中脂肪沉积,与STK25敲除后模型表现相反[31-32]。为干预疾病的进程,靶向运输STK25反义寡核苷酸(STK25 antisense oligonucleotide,STK25 ASO),发现其可选择性与肝细胞表面大量表达的去唾液酸糖蛋白受体(asialoglycoprotein receptor,ASGPR)结合,抵抗肝细胞氧化应激,改善线粒体功能,并可抑制肝脏中成脂基因及乙酰辅酶A羧化酶(Acetyl CoA carboxylase,ACC)的表达,最终延缓NAFLD的发展[33]。因此,有理由推测STK25可能通过干预脂肪酸从头合成的酶活性及氧化过程,抑制脂肪酸的相关代谢。综上所述,STK25与葡萄糖、脂质的代谢密切相关,病理状态下STK25表达上升,诱导胰岛素抵抗、糖耐量下降、脂质沉积。这一系列代谢的改变将进一步引发细胞形态和功能改变,诱导细胞的增殖或者死亡。
4 STK25与肿瘤的关系
4.1 STK25抑制Warburg效应与肿瘤
近年来诸多研究证实,肿瘤的生物学本质是一种代谢性疾病,因此基于肿瘤代谢靶点的治疗成为肿瘤治疗研究的新方向,也就是说通过调控三大基本营养物质的代谢抑制肿瘤的发生与发展。Warburg效应又称有氧糖酵解,即在含氧量正常的情况下,细胞的葡萄糖摄取量和乳酸的堆积量也会升高,利用糖酵解作为主要能量代谢的来源,获得更高的糖分解能力,这也是肿瘤细胞一个典型的代谢特征,而阻断或降低该特异性过程,可能提高抗肿瘤治疗的效果[34-35]。在一项针对结直肠肿瘤细胞增殖的研究中发现,STK25与经典的癌原蛋白Golph3均可表达于高尔基体中,并在转录及翻译层面发现,STK25的过表达显著抑制了Golph3的表达[9]。该项研究还发现STK25、Golph3的改变影响了mTORC1/mTORC2复合物的磷酸化及下游糖酵解过程。从而得出结论,STK25可通过下调Golph3依赖性mTOR途径,抑制糖酵解从而负调控细胞增殖,提示STK25可能成为抑制肿瘤细胞生长增殖的靶点。
4.2 STK25诱导细胞凋亡与肿瘤
在神经源性肿瘤中,STK25的表达对应着不同的预后水平。神经营养素受体Trk家族是神经系统细胞存活的关键调控因子,TrkA在小儿神经源性肿瘤的研究中被发现可诱导肿瘤细胞死亡,高水平的TrkA提示更好的预后[36]。研究同时发现,CCM2作为CCM3的同源物,其N端的磷酪氨酸结构域(phosphotyrosine binding,PTB)可结合TrkA,C端的Karet结构域可结合STK25,最终形成CCM2/TrkA/STK25依赖的细胞死亡途径,该途径最终仍是通过激活经典的caspase完成,因此CCM2/TrkA/STK25的作用类似于上游的开关信号[10]。也就是说,神经母细胞瘤中STK25的表达水平与CCM2和TrkA的表达水平相关,高表达STK25的患者对应更为良好的预后。
4.3 STK25负调控癌基因转录与肿瘤
除以上述2条STK25相关信号通路外,学者还发现传统的Hippo信号通路也与STK25有一定关系。Hippo信号通路在调控细胞增殖、凋亡中发挥着重要作用,该通路中关键介质是LATS1和LATS2激酶,其功能是负调控癌基因转录共激活因子YAP和具有PDZ结合基序的转录共激活因子TAZ的活性[37]。LATS1/2降解使得YAP/TAZ过度活化,在小鼠模型中促进肿瘤的发生,该现象也发生在人类多种恶性肿瘤中[38-40]。研究[41-42]已证实,Hippo通路中的关键介质LATS受MST1/2调控,MST1/2在疏水基处将LATS1/2磷酸化,去除LATS1/2的自抑制构象,从而使LATS1/2的激活环基发生自磷酸化和反磷酸化相互作用。正是这种磷酸化激活循环,从而达到完全LATS激酶活性及抗肿瘤效应。然而,在敲除MST1/2基因后,LATS信号并未完全阻断,提示一定存在其他信号分子可部分激活LATS激酶,如MAP4Ks[43]。最新研究发现,与MST1/2同家族的STK25同样可以促进LATS激活环基的磷酸化,且与之前的疏水基磷酸化无关[44-45]。该研究还指出,完全不表达STK25导致LATS活性部分降低和YAP/TAZ活性升高,肿瘤细胞增殖,而STK25 KO小鼠虽然存活,但表现出显著的肝脏肿大。即STK25为Hippo信号通路的新型调节剂,它通过独立于标准的MST/MAP4K信号通路机制激活LATS1/2,从而解释了为什么MST/MAP4Ks无法完全补偿继STK25缺失后的YAP/TAZ激活。另外,根据人类癌症数据库发现,STK25的局灶性缺失在人类癌症中很常见,如宫颈鳞状细胞癌、膀胱尿路上皮癌、头颈部鳞状细胞癌等[44]。而该局灶性丢失是否会解除Hippo信号通路,从而促进肿瘤的发生与发展有待进一步研究。
5 总结与展望
Ste20家族的生物学效应具有重要意义,其中PAK与肿瘤的增殖、侵袭密切相关,也得到了学者的广泛研究,而GCK与肿瘤之间的关系尚不明确。STK25作为GCK亚家族的重要成员之一,在慢性代谢性疾病中的研究较为成熟,如糖尿病、脂肪性肝炎等,且该分子在肿瘤发生与发展过程中也起到一定的辅助作用。但如何从代谢层面上解释与肿瘤关系的研究有待进一步深入,是否可以通过调控STK25的水平调节肿瘤糖脂代谢过程,最终抑制肿瘤的发生与发展可能成为肿瘤代谢治疗研究的一个新方向。
猜你喜欢
江苏农业科学(2022年17期)2022-09-24 11:42:26
中国医学创新(2022年18期)2022-07-12 20:01:07
天津医科大学学报(2021年3期)2021-07-21 09:03:46
四川大学学报(自然科学版)(2021年4期)2021-07-15 09:10:48
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
中国农业科学(2020年5期)2020-03-30 02:02:46
中国当代医药(2018年24期)2018-11-28 11:31:06
中风与神经疾病杂志(2017年5期)2017-01-13 03:48:36
上海师范大学学报·自然科学版(2016年5期)2016-12-14 06:31:20
中国医药生物技术(2015年4期)2015-12-26 08:26:36
