
锌稳态失衡与阿尔茨海默症
2020-08-07 07:53贺仕清唐小卿赵剑锋
中国药理学通报 2020年8期
弓 慧,贺仕清,唐小卿,赵剑锋
(南华大学 1.衡阳医学院生理学教研室、2.附属南华医院神经外科,湖南 衡阳 421001)
阿尔茨海默症(Alzheimer’s disease,AD)是一种主要累及老年人的进行性神经退行性疾病,是痴呆症最主要的病因,其临床症状主要表现为认知功能下降、精神症状和日常生活能力的逐渐下降,还伴随抑郁症、精神病等多种并发症。AD起因难寻,与家族史、甲状腺疾病和头部外伤等多种因素相关,一般根据认知能力和身体机能的恶化程度分为轻度、中度和重度痴呆3个时期。AD发病机制错综复杂,目前尚无完全行之有效的治疗手段,因此,继续研究其发病机制以及寻找合适的治疗方法仍十分重要。
AD最主要的2个病理特征是淀粉样蛋白沉积和神经纤维缠结。淀粉样蛋白斑的主要成分是β-淀粉样蛋白(amyloid β-protein,Aβ),神经纤维缠结主要成分是过度磷酸化的Tau蛋白(p-Tau)。目前普遍认为,AD的发生发展与Aβ及p-Tau的聚集、氧化应激、炎症反应、突触可塑性的损伤以及胆碱能功能缺损等多种因素密切相关。近年来,越来越多的证据也表明AD发生过程中涉及锌稳态的失衡,锌稳态失衡通过多种途径参与AD的发生。
1 锌稳态及其调控机制
锌是300多种酶或金属蛋白的辅助因子,广泛参与到细胞分裂、免疫系统、蛋白质合成、DNA合成等多种生理功能中,是机体进行正常生命活动的必需微量元素之一。大脑是人体含锌量最高的器官,约为血清锌含量的10倍,神经突触中也含有高浓度的锌,主要集中在谷氨酸能神经突触中[1]。在中枢系统中,锌以两种方式存在:稳定的结合锌与不稳定的游离锌[1]。结合锌与蛋白质和多肽紧密结合作为蛋白质折叠的关键成分,对各种胞质酶和核酶的活性至关重要;游离锌则是存在于多种细胞器腔内,如线粒体、内质网和高尔基体等,锌在这些细胞器之间进行交换,并与许多蛋白质结合,改变它们的生物活性。在正常生理情况下,机体内游离锌的浓度维持在一定水平,称之为锌稳态。在脑内,锌浓度的激增会引起神经毒性,而锌缺乏也会造成脑部中枢神经系统畸形等多种病理变化。因此,锌浓度维持相对稳定在维持机体健康起极为重要的作用,机体必然存在维持锌稳态的调节系统。
锌稳态受到机体严格调控,主要通过三大蛋白家族[1]:1) 金属硫蛋白(metallothionein,MT),抑制胞质锌毒性;2) 锌铁调控蛋白(Zrt/Irt-related proteins,ZIP),介导锌从胞外入胞或从胞内腔室流出至胞质;3) 以及锌转运蛋白(zinc transporter,ZnT),介导锌从胞内腔室和胞质出胞。三大家族通过严格控制机体锌稳态对机体多种生理功能起调控作用。在机体中,ZIPs、ZnTs和MTs都存在多种表型,各表型定位不同,共同维持机体锌稳态。
MT在生物应激或细胞损伤情况下具有生存优势,未结合金属的MT与锌结合调节锌运输和储存起到抑制锌毒性的作用。机体MT共有4种亚型:MT-Ⅰ、MT-Ⅱ、MT-Ⅲ和MT-Ⅳ。在中枢神经系统中,MT-Ⅰ、MT-Ⅱ和MT-Ⅲ广泛表达。
ZIP的功能主要是在胞质锌浓度较低的情况下将胞外和细胞器内的锌转运至胞质起到补充胞质锌浓度的作用。目前已知人类ZIP共有14种亚型,具有锌转运功能的是ZIP1-8和ZIP14。中枢神经系统中主要存在ZIP1和ZIP3,其中ZIP1的表达远高于ZIP3,因此ZIP1被认为是神经元中摄取锌的关键因子。
ZnT与ZIP的功能刚好相反,将胞质和胞内腔室中的锌排出胞外,此外,通过ZnT运出胞外的锌一个很重要的去向是转运至突触囊泡中。迄今为止,已有10种ZnT被报道具有锌转运功能。除了ZnT-3和ZnT-8分别只在大脑和胰腺中表达以外,其他ZnT大多在机体广泛表达。在大脑中,ZnT-1、ZnT-3、ZnT-4和ZnT-6高表达。
2 锌稳态失衡与AD
锌稳态对中枢神经系统正常功能的重要性不言而喻。事实上,与锌稳态失衡有关的神经系统疾病,如学习、记忆障碍通常与富含这种金属的大脑结构的功能出现异常有关,例如海马,杏仁核和新皮质等区域。目前关于AD患者体内锌稳态失衡的方向有两种研究结果。
大量研究支持锌浓度过高导致AD发生发展,研究证实老年斑中含有大量锌[2]。也有研究支持缺锌也会导致AD发病,研究发现脑锌缺乏介导突触可塑性相关蛋白表达水平降低与认知功能障碍有关[3]。除此以外,AD患者脑内参与调节锌稳态的蛋白质水平发生了变化。AD患者血清和脑脊液中的锌水平也与正常个体存在差异。由此可知,AD患者发病过程伴随锌稳态的失衡。
3 锌稳态失衡促进AD发生发展的机制
3.1 影响Aβ生成Aβ沉积形成的淀粉样蛋白斑是AD患者最典型的病理特征之一。Aβ由淀粉样前体蛋白(amyloid precursor protein,APP)经淀粉样蛋白依赖途径水解而来。APP由粗面内质网生成,经高尔基体反面网状结构(trans Golgi network,TGN),运输至细胞膜表面,依次由α-分泌酶—γ-分泌酶水解或由β-分泌酶—γ-分泌酶水解。α-分泌酶—γ-分泌酶介导淀粉样蛋白非依赖途径生成N端胞外域sAPPα和p3肽(正常神经元),β-分泌酶—γ-分泌酶介导淀粉样蛋白依赖途径生成N端胞外域sAPPβ和Aβ(AD神经元)。锌稳态失衡通过诱导APP以淀粉样蛋白依赖途径水解参与Aβ介导的AD发病。
3.1.1锌对Aβ生成的调控作用 Aβ生成主要由β-分泌酶介导。β-分泌酶与APP在内质网、TGN和质膜上的转运很大程度上影响了Aβ的生成。因此,TGN作为其中重要的一个结构,TGN及TGN所在的高尔基体的结构和功能的正常对于APP的运输至关重要。特别的是,早期AD患者的高尔基体就已经呈破碎形态。因此,关于锌对Aβ生成的调控作用,以下将分别从锌对Aβ生成酶的调控作用和锌对高尔基体结构和功能的调控作用进行阐述。
锌对Aβ生成酶存在调控作用。β-分泌酶介导的Aβ生成的整个过程涉及3个关键点:APP、β-分泌酶和γ-分泌酶。首先,锌与APP高亲和力结合,Bush等[4]发现在APP上存在高度保守的锌结合位点,促进锌与APP高亲和力结合。其次,β-分泌酶—γ-分泌酶切割APP途径中会产生一个中间物质APP-C99,锌可与APP-C99的组氨酸残基(His-6,His-13和His-14)结合而增强APP-C99二聚化,抑制γ-分泌酶对其的水解[5]。
锌对高尔基体结构和功能的完整性存在调控作用[6]。首先,Aβ可以通过破坏GRASP65-GM130复合体(高尔基体堆叠结构)导致高尔基体破碎;其次,高尔基体破碎可强烈减缓APP的运输,APP运输受阻导致淀粉样蛋白依赖机制的进行,最后产生Aβ。由此可知,高尔基体结构破坏导致的功能受损与Aβ的生成密切相关。而锌大量存在于高尔基体内,参与维持高尔基体的结构和功能的完整性。本课题组前期研究发现[7],锌分别与GRASP55 的His18 和Cys103 以及Golgin45 的Cys393 和Cys396 形成配位键,直接参与了GRASP55-Golgin45复合体(高尔基体堆叠结构)的形成;另一方面,高尔基体上存在多种锌稳态调控蛋白,主要包括ZnT-5,ZnT-6和ZnT-7以及ZIP-9和ZIP-13[8]。特别的是,ZnT6特异性位于TGN上,参与TGN结构的稳定[9]。以上发现表明锌对高尔基体结构的必要性。另外,高尔基体正常功能的执行也需要锌。高尔基体介导多种早期分泌途径,锌在早期分泌途径中起关键作用,多种ZnT和ZIP参与调节此途径[8]。
3.1.2高锌对Aβ生成的促进作用 关于锌稳态失衡对Aβ生成的影响,大多数研究支持高锌对Aβ生成具有促进作用。一方面,高锌可加速β-分泌酶介导的淀粉样蛋白依赖途径的进行。研究表明高锌可促进APP和β-分泌酶增多,进而促进Aβ生成[10]。而使用锌螯合剂处理APP转基因小鼠,发现β-分泌酶的表达和Aβ的生成均呈降低趋势[10]。因此,针对高锌对Aβ生成酶的促进作用,靶向降低锌浓度抑制β-分泌酶表达减少Aβ生成有望作为一种治疗思路。
另一方面,位于高尔基体上的锌转运蛋白表达失调也会引起锌的增加,并可能参与Aβ的生成。Lovell等[9]在AD患者的神经元中观察到了ZnT-6的增加,ZnT-6增加引起细胞内的锌在TGN中积累,使TGN破碎,并改变正常的蛋白质和脂类的分拣和转运,导致神经元退化。虽没有直接证据证明ZnT-6的增加与Aβ生成有直接关系,但鉴于TGN在APP运输中起到的重要作用,以及高尔基体破碎对β-分泌酶介导的APP水解的促进作用,推测AD患者Aβ的产生与高锌介导的TGN的破碎以及APP水解途径的改变有关。
3.1.3低锌对Aβ生成的促进作用 锌浓度过高使高尔基体破碎,锌浓度过低也可使高尔基体破碎。我们最近的研究发现低锌对Aβ生成也具有促进作用。上文提到,锌参与了GRASP55-Golgin45复合体的形成,由此参与高尔基体的正常堆叠。本课题组研究发现螯合锌可破坏GRASP55-Golgin45复合体结构,并诱导Aβ产生[11]。同样,高尔基体破碎与APP运输密切相关,那是否也可以从高尔基体破碎导致APP水解由β分泌酶介导这一角度来解释缺锌诱导的Aβ生成,也有待进一步挖掘。
综上所述,Aβ生成与锌稳态失衡密切相关,高锌或低锌都可促进Aβ生成,从而加重AD的病理症状,而恢复锌稳态可起到改善作用。
3.2 影响Aβ聚集
3.2.1锌与Aβ存在相互作用位点 锌稳态失衡除可促进Aβ生成外,也可与Aβ直接作用。Alies等[12]发现锌与Aβ 的His13、Glu11、His6 形成配位键,参与了Aβ的聚集。Khatua等[13]发现锌通过两个关键疏水链段hp1和hp2之间的远距离接触稳定Aβ肽N-末端螺旋结构,从而与Aβ高度特异性结合。锌与Aβ的大量结合,形成溶解性极低的淀粉样蛋白斑,并阻止Aβ降解[2]。
3.2.2高锌对Aβ聚集的促进作用 鉴于AD中Aβ表达呈增加趋势,而淀粉样蛋白斑的主要成分除了Aβ外,还含有大量的锌。研究多集中在探讨高锌是否可以促进Aβ聚集。研究发现高锌可使Aβ单体不稳定,并加快其聚合速度(约40倍)[14]。利用锌螯合剂[15]可释放与胞外Aβ结合的锌,恢复AD小鼠锌浓度至正常水平,并改善AD小鼠认知能力。
综上所述,对于Aβ聚集来说,高锌是一个不利因素,高锌促进Aβ聚集,导致Aβ降解困难,低锌使得锌从Aβ中释放出来,有利于AD中Aβ病理的改善。
3.3 影响Tau蛋白的聚集和磷酸化除Aβ外,AD患者最典型的病理特征即为p-Tau聚集形成的神经元纤维缠结。正常情况下,Tau蛋白作为轴突蛋白,其作用在于与微管蛋白结合促进微管形成并维持微管稳定。但在AD患者的神经元内,Tau蛋白过度磷酸化并聚集在胞质,与微管蛋白的结合能力减弱,使神经元微管结构广泛破坏,最终导致正常的轴突转运受损,引起突触丢失和神经元功能损伤。与Aβ类似,锌也与Tau蛋白存在相互作用位点并参与调节Tau蛋白的聚集和磷酸化过程。
3.3.1锌与Tau蛋白存在相互作用位点 锌与Tau蛋白存在相互作用位点。Roman等[16]发现锌与Tau蛋白存在1个高亲和力结合位点和3个低亲和力结合位点。Li等[17]发现Tau蛋白的半胱氨酸残基与锌存在结合位点。
3.3.2高锌对Tau蛋白聚集和磷酸化的促进作用 鉴于AD患者p-Tau的病理表现,目前的研究多集中在研究高锌对于Tau蛋白聚集或磷酸化的影响。Roman等[16]发现高锌可诱导Tau蛋白低聚化,但得到的低聚物是非淀粉样的,在锌螯合剂下可迅速解离。Li等[17]发现高锌可诱导Tau蛋白聚集并产生神经毒性。
还有研究表明高锌可诱导Tau蛋白磷酸化。高锌通过激活Tau蛋白磷酸化激酶促进Tau蛋白过度磷酸化,利用锌螯合剂可逆转此过程[18]。Craven等[19]发现在Tau蛋白过表达的小鼠上,补充锌增加了Tau蛋白的磷酸化水平,并进一步恶化小鼠的认知能力。
综上可知,高锌可促进Tau蛋白的聚集及磷酸化,低锌有助于改善p-Tau的病理表现。
3.4 影响突触可塑性突触可塑性指突触的形态和功能在神经元持续活动下发生较为持久的变化现象,主要作用在于维持神经元和神经环路稳定,与学习记忆形成密切相关。突触可塑性损伤可导致认知功能障碍,在AD发病中起重要作用。突触可塑性主要体现在突触结构和传递效能的变化,而这两者的结构基础大多涉及突触或神经元部位的蛋白质、神经递质和离子的生物学变化。突触锌主要位于谷氨酸能突触中,并以此与神经传递动态关联,并参与调节突触可塑性。
3.4.1锌对突触可塑性的调控作用 首先,突触结构和功能的完整性都需要锌的参与[3]。对突触结构来说,突触锌浓度维持相对稳定在突触后致密物形成中发挥重要作用,并因此参与树突棘的形成。对突触功能来说,突触锌大量存储于谷氨酸能突触末端的囊泡中,在神经元活动时与谷氨酸协同释放,是谷氨酸能神经传递的关键因子。
其次,神经营养因子发挥作用也需要锌的参与[20]。神经营养因子是一类神经元生长、存活和可塑性所必需的蛋白质分子,与突触可塑性密切相关。锌的存在对神经营养信号产生积极影响,特别是对脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)-TrkB轴的激活[3,21]。成熟的BDNF由其前体pro-BDNF在基质金属蛋白酶(matrix metalloproteinase,MMP)等胞内外蛋白酶的催化下裂解形成。MMP是细胞外基质代谢的重要酶类,通过对细胞外基质的降解影响树突棘和突触的重塑。锌作为MMP的辅助因子参与BDNF成熟[21]。这些研究表明锌的存在对突触可塑性有重要调控作用。
3.4.2低锌对突触可塑性的损伤作用 由于锌直接参与BDNF的成熟,锌依赖的BDNF成熟受损对突触结构及功能有显著影响,Zn-MMP-BDNF轴异常可损伤突触可塑性[21]。目前的研究多集中在探讨低锌对突触可塑性是否有损伤作用。Frazzini等[3]发现缺锌会抑制BDNF成熟,并且损伤年轻小鼠的认知功能。而补充锌对BDNF-TrkB轴有激活作用并可改善年轻小鼠的认知功能[3],也可恢复AD小鼠BDNF表达并改善小鼠认知损伤[22]。
3.4.3高锌对突触可塑性的损伤作用 虽然突触可塑性的形成需要一定浓度的锌,但锌浓度过高也会带来副作用。Shetty等[23]发现老年大鼠海马区锌浓度增加,导致迟发性长时程增强介导突触可塑性损伤,利用锌螯合剂可恢复受损的突触可塑性。
综上所述,高锌或低锌均可诱导突触可塑性损伤,靶向恢复锌稳态可改善AD的突触可塑性损伤。
3.5 影响氧化应激氧化应激指体内氧化与抗氧化作用失衡,产生大量自由基,主要包括氧自由基(reactive oxygen species,ROS)与氮自由基(reactive nitrogen species,NOS)。目前普遍认为氧化应激是AD疾病进程的早期和持续事件。AD患者脑内显示出明显的蛋白质、脂质和核酸的氧化损伤,在淀粉样蛋白斑块周围,氧化损伤也高度集中甚至蔓延至无Aβ沉积的神经纤维中。锌稳态失衡通过促进氧化应激参与到加速Aβ的沉积并加速AD的进程。
3.5.1锌对氧化还原代谢的调控作用 锌在生物体内具有氧化还原惰性,但对氧化还原代谢有重要影响。首先,锌具有良好的细胞保护作用,主要体现在两方面:1) 保护细胞不受氧化损伤,特别是保护细胞巯基不受氧化和抑制过渡金属产生活性氧;2) 保护蛋白质和核酸不被氧化和降解,同时稳定微管细胞骨架和细胞膜[24]。其次,锌虽然本身对氧化还原不敏感,但由于MT具有很高的金属结合能力,又具有很强的氧化还原能力(MT含巯基侧链,MT-Ⅲ的巯基侧链氧化还原电位很低(-366 mV),极易被氧化[25]),这使得与MT结合的锌对氧化还原变得敏感,也使得这种状态下的锌对氧化应激十分敏感。

因此,胞质锌浓度升高可通过多种渠道引起氧化应激增强,并导致细胞毒性。
3.6 高锌对胆碱能功能缺损以及炎症反应的促进作用除上述所述机制以外,还有多种机制也参与了AD的发生,而这些或多或少都与锌稳态失衡有关。例如胆碱能功能缺损、炎症反应等。
胆碱能功能缺损主要体现在乙酰胆碱能代谢活动下降,例如胆碱乙酰转移酶(choline acetyltransferase,ChAT)活性降低[26]。Tabrizia等[26]发现口服氯化锌可降低小鼠ChAT蛋白表达,并引起认知功能损伤。另外,谷氨酸能突触中过量的锌具有神经毒性,会导致能量平衡的崩溃和胆碱能神经元功能和结构的损伤[27]。
AD患者也存在炎症反应。Aβ作为一种炎症刺激因子,可以激活小胶质细胞和星形胶质细胞释放具有强烈毒性的炎症因子[28]。Higashi等[29]发现当胞外锌大量增加时,小胶质细胞被激活,产生大量促炎性细胞因子。
以上两点均说明,对于胆碱能功能和炎症来说,高锌是一个危险因素。但锌本身在其中起到什么作用,还有待进一步研究。
4 锌稳态调控蛋白表达失调与AD发生相关
上文表明锌稳态失衡与AD发病机制密切相关,作为起主要调控作用的MT、ZIP和ZnT,它们的表达失调也同样与AD的发生发展密切相关,Tab 1展示了研究较多的与AD相关三大蛋白家族在大脑中的定位、主要作用以及在AD中的变化趋势。
Tab 1表明,AD的发生伴随着锌稳态相关调控蛋白的失调。研究人员也针对它们做了一系列与AD发病相关的研究。针对MT,目前普遍认为MT-Ⅲ是治疗AD的一个潜在靶点。持续性注入Zn7MT-Ⅲ至中枢神经系统后,Aβ斑块面积减少,ROS产生减少,AD小鼠认知能力明显改善[30]。通过药物增加MT-Ⅲ水平有望是治疗AD的一个重要方向。针对ZnT,目前研究最多的是ZnT-3。多项研究发现敲除ZnT-3可降低突触中的锌浓度,进一步使BDNF,pro-BDNF等与突触相关蛋白减少,并导致老年小鼠的认知功能出现损伤,但靶向ZnT-3表达下降进行治疗的方案有待进一步研究[21]。针对ZIP,研究发现在AD果蝇模型中,敲除ZIP1可增强果蝇的攀爬能力,也可延长果蝇的寿命[20]。由此可见,锌稳态调节蛋白表达异常与AD发展相关,靶向恢复特定锌稳态调控蛋白的表达可改善AD病理及认知损伤。
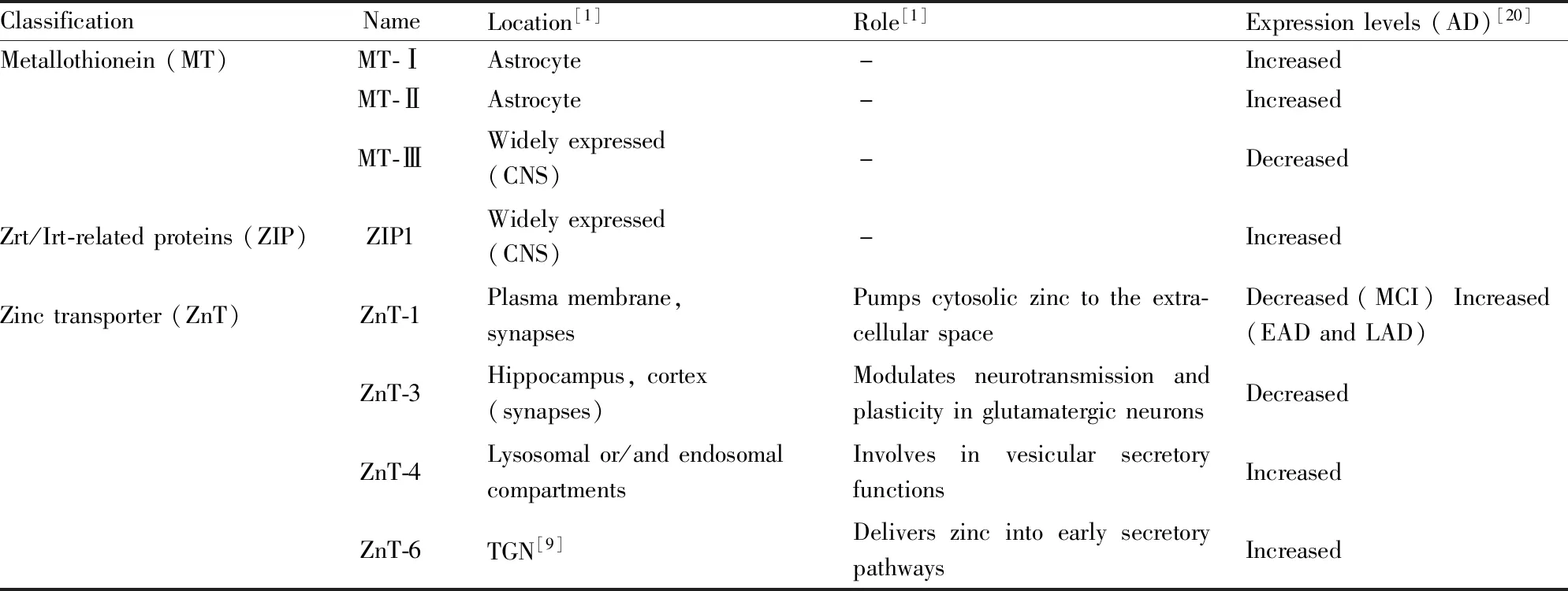
Tab 1 Location and role of brain zinc homeostasis regulatory protein and their alterations in Alzheimer’s disease
5 结语
目前研究发现锌与AD密切相关的Aβ,p-Tau以及神经突触的功能及作用存在调控作用,锌稳态失衡通过加速Aβ的生成和聚集、加速Tau蛋白的聚集和磷酸化,以及损伤突触可塑性参与AD的发生和发展,而针对各自不同机制进行靶向恢复锌稳态可有效改善AD病理及认知损伤。其具体调节锌稳态作用的MT、ZIP和ZnT蛋白也被发现在AD中发生了变化,靶向特定蛋白进行恢复也可以起到改善AD病理及认知损伤的作用。因此,锌稳态失衡与AD发生发展密切相关,是AD的关键机制,而靶向恢复锌稳态,无疑给AD患者的治疗带来无限曙光。
目前有一些针对恢复锌稳态的药物正在研究中,比如氯碘羟喹(clioquinol,CQ)(锌螯合剂)。研究表明,CQ可透过血脑屏障,并抑制Aβ沉积和ROS产生;二期临床试验中,中重度AD患者口服CQ后,记忆衰退的速度变慢,血浆Aβ42水平下降[24]。另外,CQ的衍生物PBT-2也又类似的作用,且比CQ拥有更加广阔的研究前景[20-21]。
然而,迄今为止,锌稳态失衡导致AD发生发展的一些问题尚待回答,例如锌稳态失衡导致Aβ沉积等AD相关病理改变是否存在关键中间因子?此外,锌稳态调节蛋白表达异常与AD发生发展还存在很多未解之谜。总之,锌稳态失衡在AD等神经退行性疾病发生发展的作用已引起广泛关注,越来越多的研究也在尝试深入探讨其分子机制,从而为靶向锌稳态失衡防治AD提供更多更加充分的科学依据。
猜你喜欢
大电机技术(2022年3期)2022-08-06
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2022年1期)2022-02-28
核科学与工程(2021年4期)2022-01-12
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年6期)2021-07-31
煤气与热力(2021年4期)2021-06-09
中华戏曲(2020年1期)2020-02-12
分析化学(2017年12期)2017-12-25
