
多酶催化串联策略在复杂天然产物合成中的应用
2020-08-07 05:53:06贺俊斌孟松潘海学唐功利
合成生物学 2020年2期
贺俊斌,孟松,潘海学,唐功利
(中国科学院上海有机化学研究所,生命有机化学国家重点实验室,上海200032)
结构新颖、复杂多样的天然产物及其衍生物一直以来都是现代药物的重要组成部分和新药发现的重要源泉。研究表明,在1981—2014 年间上市的1500 多种小分子药物中,超过50%的药物直接或间接来源于天然产物及其衍生物,包括著名的抗疟药青蒿素和抗肿瘤药紫杉醇[1]。然而,传统的从植物、微生物及动物中提取分离天然产物的方法往往存在步骤繁琐、耗时长、成本高、提取效率低等问题,一定程度上制约了新型药物的研发[2-3]。因此,人工合成具有重要生物活性、结构复杂、新颖的天然产物具有重要意义。
天然产物全合成主要包括化学合成和生物合成。自1828 年德国化学家Wöhler 实现尿素的合成以来[4],天然产物的化学全合成取得了辉煌的成就,化学家们不断挑战合成结构复杂的天然产物,完成了包括维生素B12、海葵毒素、软海绵素等一系列具有里程碑意义的全合成工作[5-9]。尽管人们能够设计合成自然界存在的几乎任何天然化合物,甚至可以合成理论存在但自然界尚未发现的设想的理论分子,但在一些含有多手性中心、结构复杂的天然产物化学全合成过程中也存在一些问题,如合成步骤繁琐(通常几十步)、产率低(通常<1%)、产物选择性不强、反应条件剧烈、需要昂贵试剂或所用有机溶剂易造成污染、难以实现大量合成等[10-11]。近年来,随着高通量测序技术、生物信息学、分子生物学等技术的发展,天然产物生物合成取得了巨大的进展。以合成生物学、酶催化合成、组合生物合成为主的生物合成为复杂天然产物的全合成提供了有效策略[12-16]。其中,体外酶催化合成因其具有催化效率高、反应选择性强、产物专一性强、反应条件温和、绿色环保等优点[17-20],吸引了研究人员的关注。本文作者介绍了近年来采用体外酶催化方法合成复杂天然产物的相关研究进展,重点介绍了多酶催化串联法在复杂天然产物合成中的应用研究进展以及目前存在的问题和解决对策。
1 多酶催化串联法合成复杂天然产物
串联反应提供了一种新的合成思路,其体系是一套经过精心设计的反应系统,每一步反应的产物都作为下一步反应的底物,在整个反应体系中初始底物经过逐步催化后,会最终转化为结构复杂的功能分子。在复杂天然产物的全合成中设计串联反应体系具有可以节省原料、溶剂及催化剂用量、减少反应时间、无需进行每步反应之间的分离纯化步骤等众多优点[21]。因此,串联反应体系已成为天然产物全合成中的重要合成策略。
酶催化串联反应源于大自然中生命有机体为了维持生命过程而进化出的各种代谢通路。本质上,大自然中的生命体便是一个复杂精妙的一锅多酶串联体系,在这个串联系统中,酶作为催化剂,在多个区间分别参与进行不同的代谢合成反应,多步反应环环相扣,最终形成结构复杂、多种多样的生物活性分子(天然产物)[22-24]。体外酶催化串联反应可以分为一锅串联、一锅分步串联以及分步串联。一锅串联反应步骤简单、无需分离中间体产物、合成效率高,但有时体系中存在的辅因子或产生的副产物可抑制某些酶的活性。对此,可采用一锅分步串联或分步串联进行解决。然而,一锅分步串联步骤相对繁琐,需要目标中间体产物产生后,才能进行下一步串联反应。此外,分步串联需要中间体产物的分离纯化,且耗时较长。但不管哪种串联方式,相比于化学催化剂,酶作为催化剂的多酶催化串联体系往往具有催化效率高、毒性小、反应条件简单温和以及容易控制、选择性强(化学、立体和区域)、产物构型专一、省时环保等特点,因而在药物、精细化工品、食品、农化产品等的合成生产中得到了广泛的应用[17-19,23,25-26]。随着测序技术、生物信息学、分子生物学、遗传操作等技术的发展,近年来越来越多结构复杂的天然产物的生物合成途径得到解析,在此基础上研究者们也采用多酶催化串联法成功合成了许多具有重要活性的天然产物。本节就近年来多酶催化串联策略在复杂天然产物合成中的应用作一介绍。
1.1 聚酮类化合物
肠道菌素(enterocin,1)是由日本科学家于1975年分离于链霉菌(Streptomycessp.)的一种聚酮类抗生素,具有良好的抗菌活性[27]。随后人们从海洋微生物S.maritimus中也分离得到肠道菌素(1)以及聚酮类化合物wailupemycins[28]。Moore课题组[28-30]一直从事肠道菌素(1)的生物合成途径、体外酶催化合成以及类似物合成研究,通过异源表达生物合成基因簇、基因敲除以及体外酶催化实验成功解析了肠道菌素(1)的生物合成途径。肠道菌素(1)的生物合成来源于1 分子的苯甲酸(benzoic acid)和7 分子的丙二酸单酰辅酶A(malonyl-CoA),其生物合成基因簇中并不包括编码环化酶或芳香化酶的基因,而是包含一个编码黄素依赖的氧化酶基因(encM),该基因负责催化类Favorskii 氧化重排反应从而形成肠道菌素(1)独特的笼状三环和吡喃酮结构。在解析肠道菌素(1)的生物合成途径后,该课题组通过表达纯化肠道菌素(1)生物合成途径中的关键酶以及利用商业化的辅因子蛋白,采用一锅分步串联在体外成功合成了肠道菌素(1),其总产率约25%[31]。值得一提的是,在体外一锅串联EncA/B、EncC、EncD 和EncN 蛋白时,其产物主要以wailupemycin F(2)和wailupemycin G(3)为主。随后该课题组又通过不同酶相互串联以及以苯甲酸类似物为底物,成功在体外酶法全合成了8个5-deoxyenterocin(4)类似物和16 个wailupemycin 类似物,其中包括18 个新化合物[32]。肠道菌素(1)的生物合成、体外酶催化合成以及类似物研究为后来复杂天然产物的生物合成研究提供了很好的思路(图1)。
Gilvocarcins是S.griseoflavusGö3592 及其他链霉菌产生的一系列具有苯并[d]萘[1,2-b]吡喃-6-酮骨架结构的非典型角蒽环聚酮化合物,具有显著的抗肿瘤活性[33-34]。 腊伯罗霉素(rabelomycin,5)和defucogilvocarcin M(6)是gilvocarcin 生物合成途径中的两个中间体[35-36]。腊伯罗霉素(5)是1970 年分离于橄榄色链霉菌S.olivaceusATCC 21549 发酵液中的一种角蒽环类抗生素,具有良好的抗革兰氏阳性菌活性[37]。Rohr课题组[38]利用不同生物合成途径来源的聚酮合酶,即来源于gilvocarcin、ravidomycin 和杰多霉素(jadomycin)生物合成途径,采用多酶催化串联一锅法以乙酰辅酶A(acetyl-coA)和丙二酸单酰辅酶A 为底物在体外成功合成了抗生素腊伯罗霉素(5)。该体系包括6 个酶:酮基合成酶KSα和链长因子KSβ融合的蛋白JadAB、酰基载体蛋白质RavC、酰基载体蛋白质转酰基酶GilP、聚酮合酶相关的酮基还原酶GilF、两个环化酶RavG 和JadD,经一锅反应2h,腊伯罗霉素(5)的产率可达80%(图2)。在此基础上,该课题组又将6个不同来源的后修饰酶(3 个加氧酶GilOI、GilOII 和JadF、2 个甲基转移酶GilM 和GilMT 以及1 个氧化还原酶GilR)与上述聚酮合酶进行串联,并加入了辅因子再生所需的黄素还原酶(flavin reductase)和葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase);通过15 个酶进行串联催化,一锅法在体外成功合成了defucogilvocarcin M(6)[36]。腊伯罗霉素(5)和defucogilvocarcin M(6)的体外全酶法合成进一步阐明了gilvocarcin生物合成途径中的氧化重排反应,同时也为角蒽环聚酮化合物的全合成提供了新的策略。
司替霉素(steffimycin)是1967 年分离于斯特菲斯堡链霉菌(S.steffiburgensis)的一种蒽环类抗生素,具有显著的抑癌作用;presteffimycinone(7)是其生物合成途径中的一个关键中间体[39-41]。同样地,利用光辉霉素(mithramycin)、gilvocarcin和司替霉素生物合成途径中的不同聚酮合酶和后修饰酶,以乙酰辅酶A和丙二酸单酰辅酶A(由丙二酰辅酶A:酰基载体蛋白质转酰基酶MatB合成)为底物,通过多酶催化串联一锅法在体外成功合成了presteffimycinone(7),其对确定司替霉素生物合成途径中环化酶StfX 和酮基还原酶StfT 的催化顺序提供了新的参考(图2)[39-40]。类似地,聚酮类抗生素特曲霉素(tetracenomycins)也通过体外全酶法催化合成[42-43]。
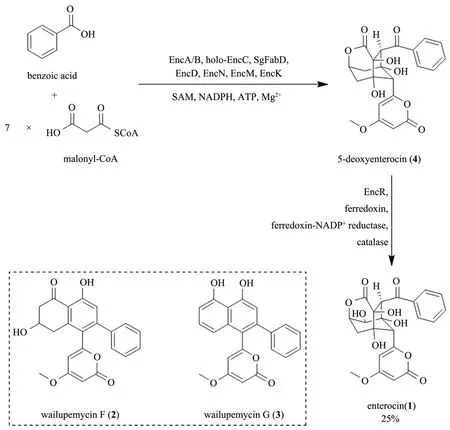
图1 多酶催化串联法合成肠道菌素(1)和wailupemycinsFig.1 Multienzyme-catalyzed tandem synthesis of enterocin(1)and wailupemycins
1.2 生物碱类化合物
赛洛西宾(psilocybin,8)又名裸头草碱、裸盖菇素、光盖菇素,最早分离于致幻蘑菇Psilocybe属,是一种具有神经致幻作用的神经毒素,结构上属于吲哚生物碱类(色胺衍生物)[44-45]。研究表明,赛洛西宾(8)具有潜在的治疗抑郁症等精神疾病以及药物上瘾的效果,目前正在进行治疗抑郁症的Ⅱ期临床试验[46-47]。尽管赛洛西宾(8)在减轻抑郁症及相关疾病方面具有潜在的应用价值,但其生物合成途径一直以来未被解析。最近,Fricke等[48]通过体外酶催化实验以及生物转化成功解析了赛洛西宾(8)的生物合成途径,其主要有4个酶参与:色氨酸脱羧酶PsiD、细胞色素P-450 单加氧酶PsiH、激酶PsiK 和甲基转移酶PsiM。进一步的体外多酶催化串联一锅法研究发现,以4-羟基-L-色氨酸(4-hydroxy-L-tryptophan)为底物,利用PsiD、PsiK和PsiM 三个酶即可在体外实现赛洛西宾(8)的酶法全合成(图3),产率约26%。赛洛西宾(8)的体外酶法全合成研究进一步体现了多酶催化串联法在药物合成中的重要应用价值。
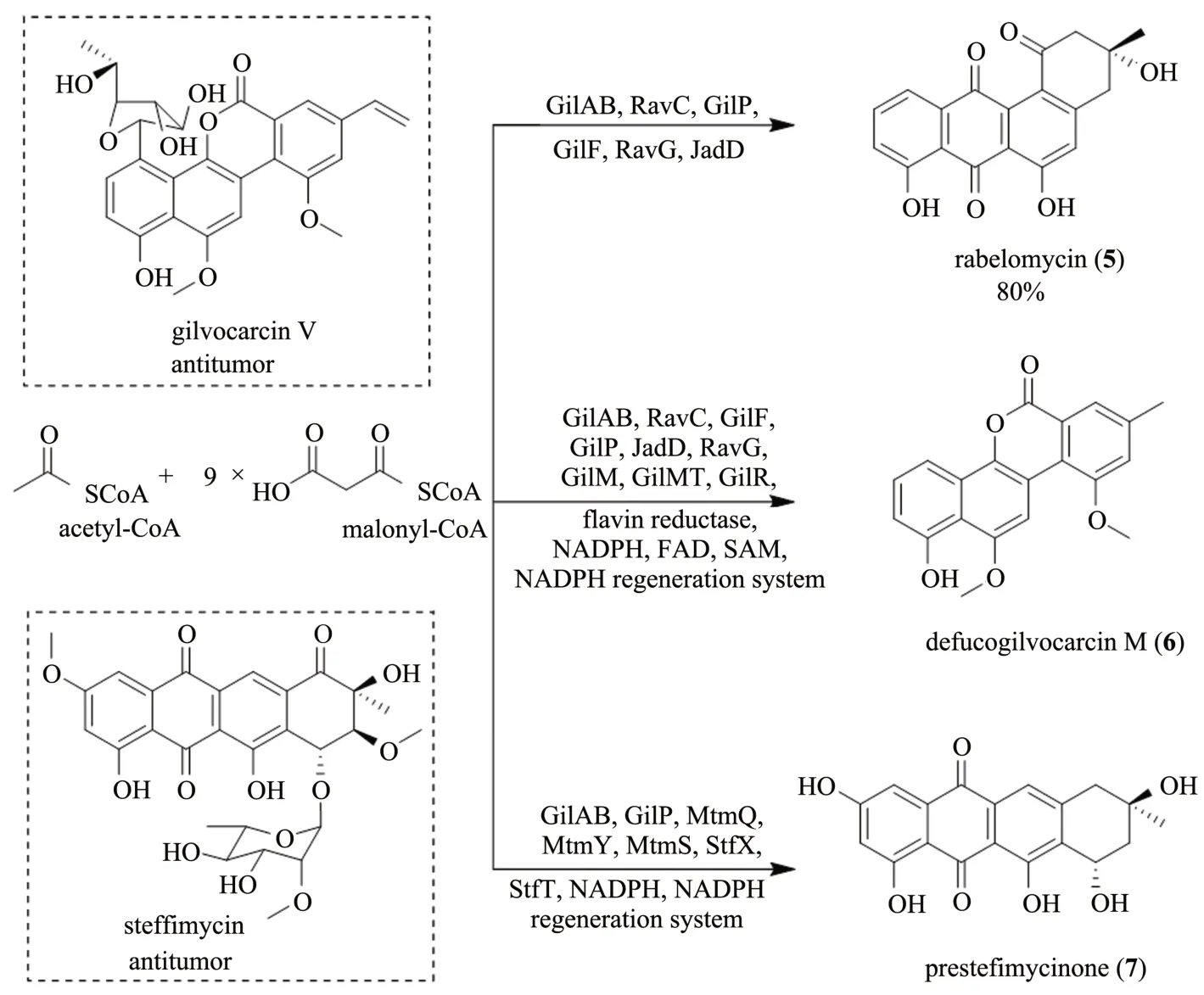
图2 Gilvocarcin V和司替霉素的结构式及多酶催化串联法合成腊伯罗霉素(5)、defucogilvocarcin M(6)和presteffimycinone(7)Fig.2 Chemical structures of gilvocarcin V and steffimycin and the multienzyme-catalyzed tandem synthesis of rabelomycin(5),defucogilvocarcin M(6)and presteffimycinone(7)
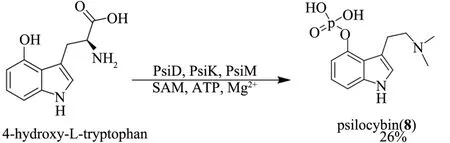
图3 多酶催化串联法合成赛洛西宾(8)Fig.3 Multienzyme-catalyzed tandem synthesis of psilocybin(8)
苄基异喹啉生物碱(benzylisoquinoline alkaloids)是一类结构多样的重要天然产物,具有抗菌、抗肿瘤、抗炎、抗氧化、抗病毒、心血管保护以及中枢神经系统等方面的药理活性,如目前广泛使用的镇痛药吗啡和可待因、抗癌药长春花碱以及具有抗菌消炎作用的小檗碱等[49-50]。苄基异喹啉生物碱目前主要从开花植物中提取获得,但存在植物资源匮乏、含量低、提取难度大、纯度低等问题;此外,化学全合成又受到产物手性的限制[49,51]。生物合成途径上,苄基异喹啉生物碱的合成一般以氨基酸作为初始前体,如酪氨酸,再经过一系列的羟化、脱羧、甲基化、Pictet-Spengler 缩合以及氧化还原等酶催化反应生成不同的化合物[52]。最近,Wang 等[53]通过多酶催化串联反应成功实现了苄基异喹啉生物碱的体外酶法全合成。研究人员首先通过克隆表达与功能验证实验,筛选获得了来源于Candidatus Nitrosopumilus salariaBD31 中的酪氨酸酶CnTYR和粪肠球菌(Enterococcus faecalisDC32)的酪氨酸脱羧酶EfTyrDC,随后将它们与来源于紫色杆菌 (Chromobacterium violaceum) 的转氨酶CvTAm 和来源于黄唐松草(Thalictrum flavum)的去甲乌药碱合成酶TfNCS 进行组合,以L-酪氨酸及其衍生物为底物,通过酶催化串联一锅法或分步串联在体外成功合成了8 个高立体选择性的苄基异喹啉生物碱[(9)~(16)],包括6 个非天然生物碱;其转化产率约35%~99%,分离产率约23%~66%(图4)。其中,化合物13 能达到克级别规模的合成,为复杂天然产物的规模化制备提供了重要例子。
Thaxtomin 是疮痂病菌代谢产生的一类植物毒素,最早是在1989 年分离于感染疮痂病的土豆切片中;thaxtomin 系列化合物是一类含有4-硝基吲哚的二酮哌嗪分子,其化学结构的显著特征是4-硝基吲哚和C-羟基-2,5-二酮哌嗪环,差别在于N-甲基、酚羟基以及二酮哌嗪环上羟基的存在与否以及取代与否[54]。因其独特的化学结构组成单元,thaxtomin 家族化合物[(17)~(21)]尤其是thaxtomin A(17)作为除草剂和挑选耐受疮痂病的土豆品种的筛选剂在农业领域具有潜在的应用价值[55]。然而在化学全合成thaxtomin 中仍存在立体或区域选择等挑战,而生物合成为其提供了更有效的策略[56-58]。Jiang 等[57]首先通过体外酶催化实验,确定了thaxtomin 生物合成途径中TxtA、TxtB和TxtC 三个酶的功能并考察了它们的底物谱,其中TxtA 和TxtB 是两个非核糖体肽合成酶,它们催化苯丙氨酸和4-硝基色氨酸合成N,N-二甲基二酮哌嗪环并具有底物宽泛性,而TxtC 是一个细胞色素P-450酶,其可以催化thaxtomin D(20)的C-14和C-20 位发生羟基化反应。随后研究人员们将这三个酶与thaxtomin 生物合成途径中负责催化L-色氨酸(L-tryptophan)C-4 位发生硝基化反应的P-450酶TxtE(嵌合蛋白TB14)进行串联,成功在体外通过酶催化全合成了thaxtomin化合物(图5)。起初将TB14、TxtA、TxtB以及TxtC进行串联,以L-色氨酸和L-苯丙氨酸(L-phenylalanine)为底物一锅法合成thaxtomin 化合物,但总转化产率低于9%,可能与反应体系中存在的NADPH再生体系有关。随后他们采用分步串联的策略,即首先串联TB14、TxtA 和TxtB 三个酶,然后再加入TxtC,thaxtomin 的转化产率可达47%。通过多酶催化串联一锅法或分步串联一锅法策略,以L-色氨酸、L-苯丙氨酸以及它们的类似物为底物,Jiang 等[57]在体外实现了124 个thaxtomin 化合物的酶催化全合成,且研究表明部分化合物具有显著的除草活性(IC50值小于2 μmol/L)。Thaxtomin化合物的体外酶法全合成进一步体现了酶作为催化剂在丰富天然产物结构多样性方面的重要作用。
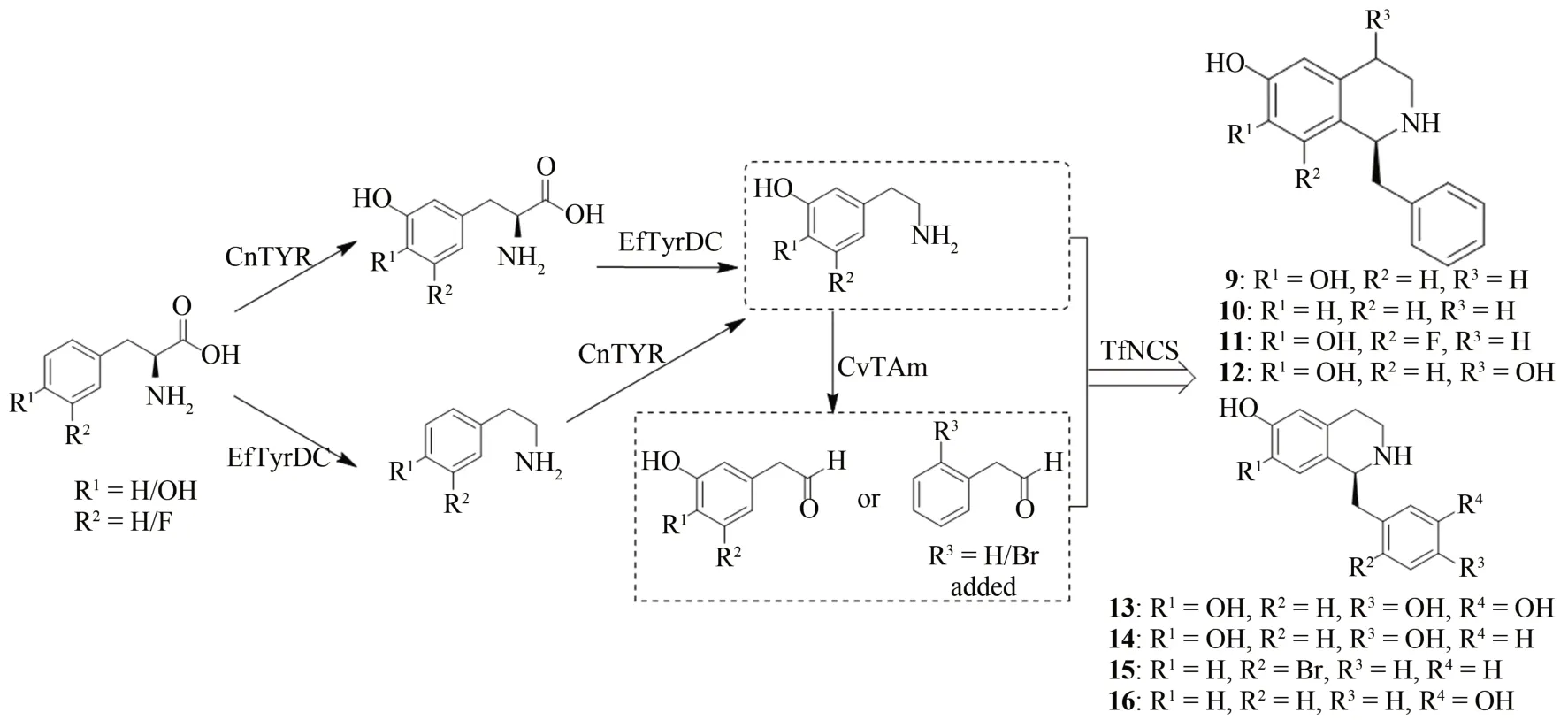
图4 多酶催化串联法合成苄基异喹啉生物碱[(9)~(16)]Fig.4 Multienzyme-catalyzed tandem synthesis of benzylisoquinoline alkaloids[(9)~(16)]
双环霉素(bicyclomycin,22)是日本科学家于1972 年从链霉菌中分离获得的一种抗生素,具有显著的抗革兰氏阴性菌活性,也是目前已知的唯一来源于天然产物的转录终止因子Rho蛋白的选择性抑制剂[59-61]。双环霉素(22)属于氧杂桥环二酮哌嗪类化合物,其结构特异,可分为三部分:[4,2,2]-氧杂桥环骨架、C1 三羟基基团以及C5=C5a环外亚甲基(图6)。作为一种具有桥环三维结构、脂肪链被高度氧化修饰的活性天然产物,双环霉素(22)自发现以来就引起了有机合成化学家的极大兴趣,其最高效且立体专一性的化学全合成是由Williams等[62-63]完成,整个合成路线有12步,总产率约为4%~5%。作者课题组一直从事复杂天然产物的生物合成研究,最近通过采用在体外重构所有酶催化反应的策略成功解析了双环霉素(22)的生物合成途径[64]。双环霉素(22)的生物合成途径包含7 个酶:1 个环二肽合酶BcmA、5 个非血红素铁依赖的双加氧酶BcmB/C/E/F/G和1个细胞色素P-450 单加氧酶BcmD。其详细途径在体外通过逐个酶催化的方法解析如下:首先,BcmA 催化一分子异亮氨酰tRNA 和一分子亮氨酰tRNA 缩合并环化,形成双环霉素(22)骨架环异亮氨酸亮氨酸(cIL);随后在BcmE、BcmC 和BcmG 催化的连续羟化反应以及BcmB催化的脱氢-环氧化作用下,形成具有[4,2,2]-双环骨架的氧杂桥环;最后经BcmD 催化的羟化反应和BcmF 催化的脱氢反应形成最终的产物双环霉素(图6)。在此基础上,以环二肽cIL 为底物,笔者课题组通过采用分步串联策略,即BcmE 首先催化cIL 发生羟化反应,随后同时加入BcmB、BcmC、BcmD、BcmF和BcmG,一锅法在体外实现了双环霉素(22)的酶法全合成。双环霉素(22)的生物合成途径中包括了连续多步惰性C—H键活化,以及在C—H键活化基础上的一步脱氢-环氧化-桥环形成过程,其体外酶法全合成的实现为构筑含有多手性中心中链氧杂桥环的分子提供了的新途径。
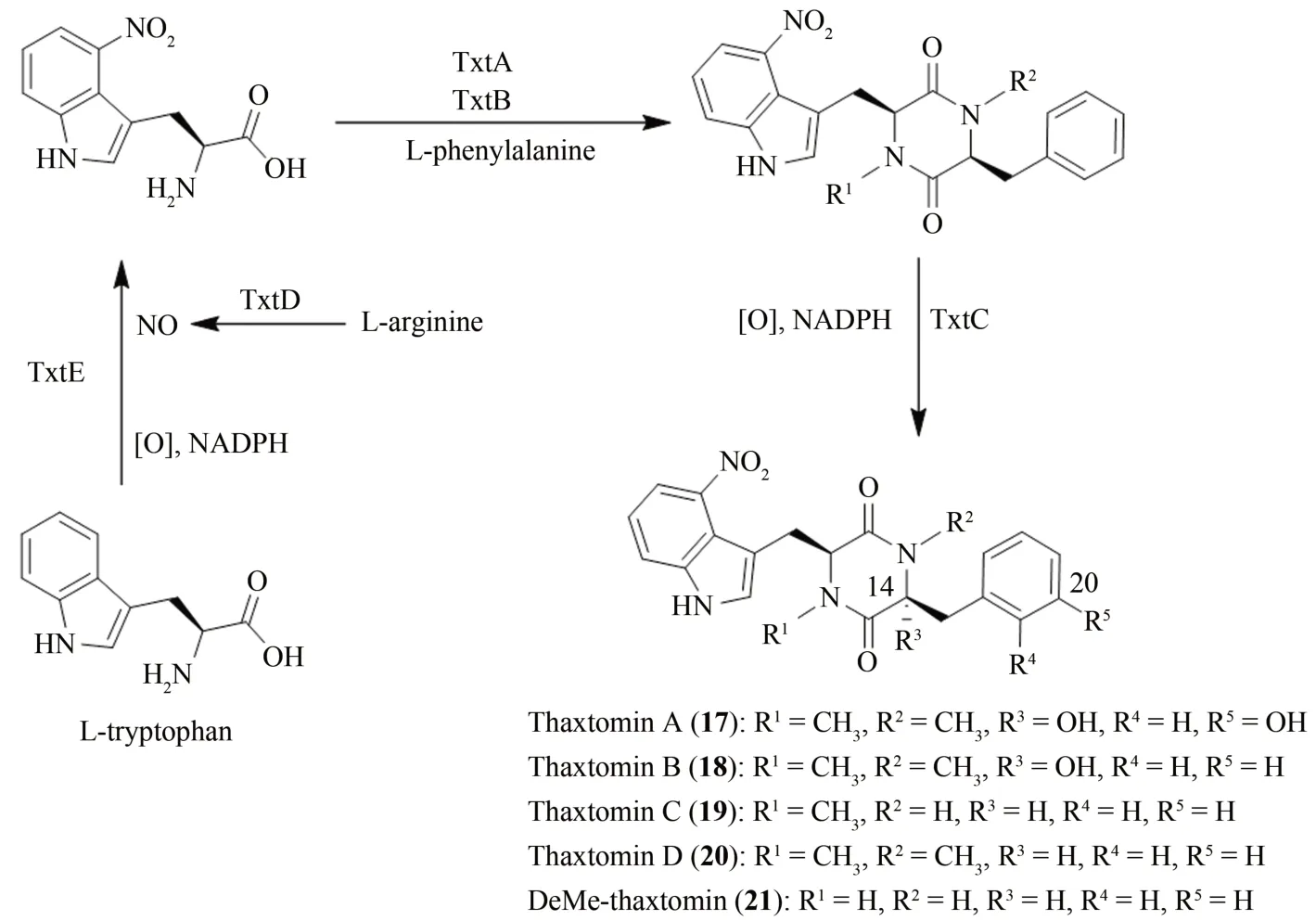
图5 Thaxtomins的生物合成途径Fig.5 Biosynthetic pathway of thaxtomins
1.3 萜类和甾体类化合物
赤霉素类(gibberellins,GAs)化合物是一类具有重要生理活性的植物激素,它们参与许多植物的生长发育过程,如种子发芽、茎干延长、诱导开花、性别分化等,目前在农业、林业、酿造业、化妆品等领域具有广泛应用[65]。赤霉素类化合物在结构上属于四环二萜,其生物合成途径目前基本被阐明。同植物其他萜类化合物的生物合成途径,赤霉素类化合物的生物合成分为3个过程:牻牛儿基牻牛儿基焦磷酸(geranylgeranyl diphosphate,GGPP)的合成,其主要是由乙酰辅酶A 通过甲羟戊酸(mevalonate)途径或甲基赤藻糖磷酸途径合成;GA12-醛的合成,其是由GGPP经古巴焦磷酸合成酶(copalyl pyrophosphate synthase,CPS)、内根-贝壳杉烯合成酶(ent-kaurene synthase,KS)、内根-贝壳杉烯酸氧化酶(ent-kaurenoic acid oxidase,KAO)以及内根-贝壳杉烯氧化酶(ent-kaurene oxidase,KO)催化合成;赤霉素类化合物的合成,包括多步的氧化以及氧化去饱和过程[65]。
2011 年,Sugai 等[66]成功实现了赤霉素GA4(23)的体外酶法全合成(图7)。研究人员首先以乙酸钠(sodium acetate)为起始底物,通过甲羟戊酸途径中4个酶的催化,在体外成功合成了甲羟戊酸,产率约54%。随后以甲羟戊酸为底物,串联6个生物合成相关的酶,成功在体外合成了关键前体化合物内根-贝壳杉烯(ent-kaurene),产率约3%;而将这10个酶串联起来也可以通过一锅法全合成内根-贝壳杉烯,但产率仅为0.3%。最后,研究人员将分离得到内根-贝壳杉烯为底物,在反应体系中加入两个P-450氧化酶KO 和KAO 以及两个非血红素铁依赖的氧化酶20ox 和3ox,成功合成了赤霉素GA4(23)。同样地,将上述14 个酶进行串联通过一锅法全合成GA4(23)时,产率为0.15%。
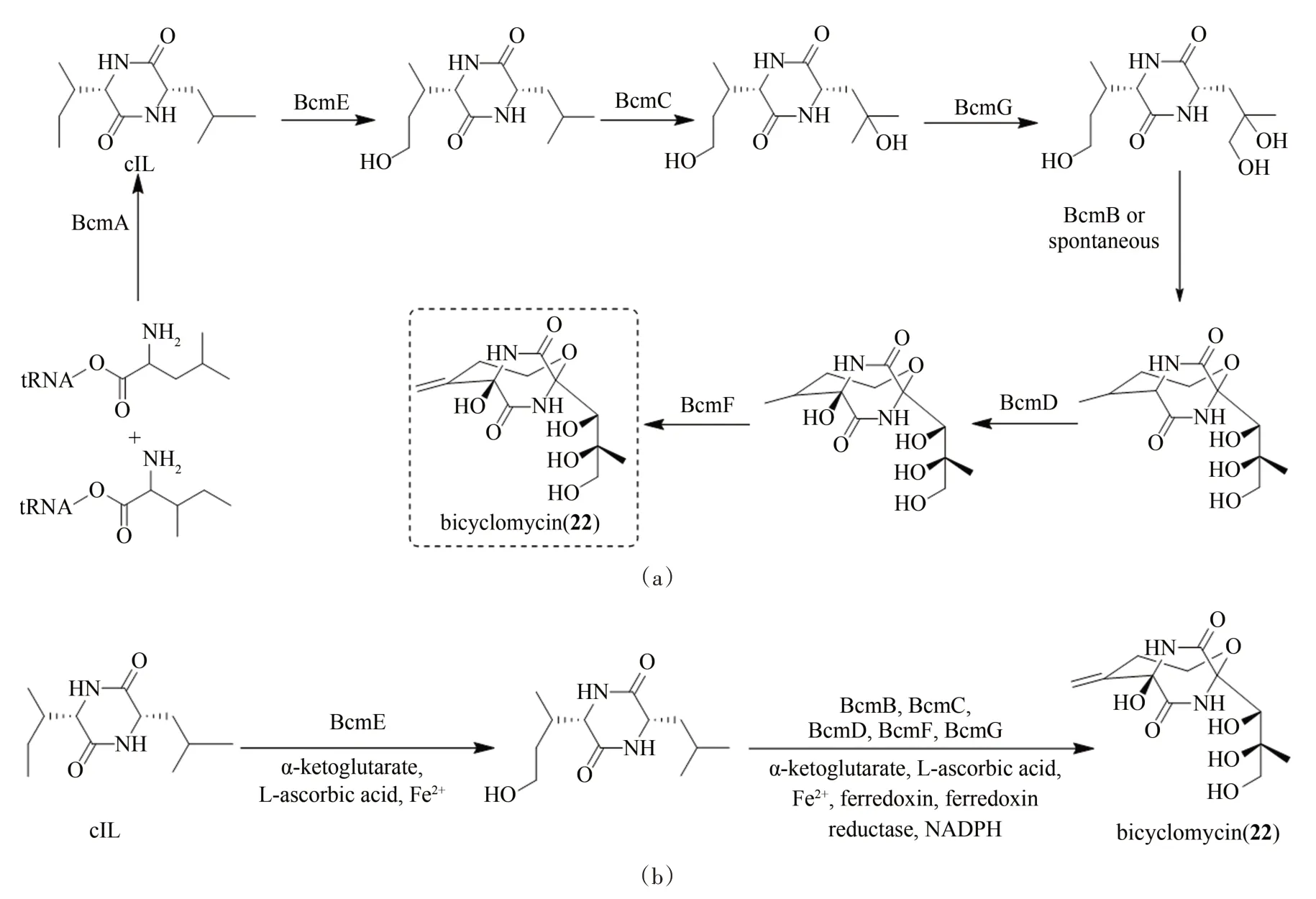
图6 双环霉素(22)的生物合成途径(a)和体外酶催化合成(b)Fig.6 Biosynthetic pathway(a)and in vitro multienzyme-catalyzed synthesis(b)of bicyclomycin(22)
熊去氧胆酸(ursodeoxycholic acid,UDCA,24)属甾体胆酸类化合物,在临床上主要用于治疗胆结石、胆汁淤积性肝病、肝炎、脂肪肝等相关疾病[67]。UDCA(24)目前主要是由牛或鹅等动物的胆汁中提取的鹅去氧胆酸(epimer chenodeoxycholic acid,CDCA)通过化学半合成获得,但其产量并不能有效满足临床需求;生物合成中UDCA(24)则是由羟甾类脱氢酶类(hydroxysteroid dehydrogenases,HSDHs)催化CDCA转化产生[68-69]。华东理工大学的许建和课题组[68]最近通过采用酶催化串联一锅法在体外成功实现了CDCA到UDCA的高效转化。研究人员首先鉴定了一个来源于扭链瘤胃球菌(Ruminococcus torquesATCC 35915)的自养型羟甾类脱氢酶7β-HSDHRt,随后将该酶与来源于Clostridium absonum的自养型羟甾类脱氢酶7α-HSDHCa进行串联,在体外一锅法实现了从CDCA到UDCA(24)的合成,产率约73%,但同时存在中间体产物7-O-石胆酸(7-oxolithocholic acid,7-oxo-LCA)约5%。为了减少体系中中间体副产物的产生,研究人员随后将7α-HSDHCa替换为来源于大肠杆菌(Escherichia coli)的7α-HSDHEc,采用分步串联一锅法进行UDCA(24)的合成,即首先由7α-HSDHEc和乳酸脱氢酶LDH进行CDCA的第一步氧化反应,待CDCA 完全转化后对7α-HSDHEc和LDH进行煮沸灭活;再加入7β-HSDHRt和葡萄糖脱氢酶GDH 进行第二步还原反应。通过分步串联一锅法,UDCA(24)的产率可高达98%(图8)。

图7 多酶催化串联法合成赤霉素GA4(23)Fig.7 Multienzyme-catalyzed tandem synthesis of gibberellin A4(23)
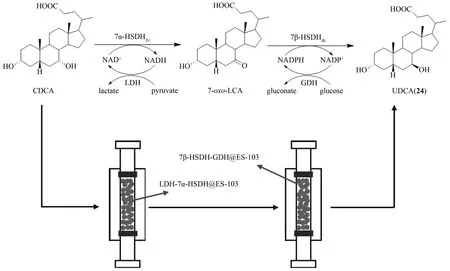
图8 多酶催化串联法合成熊去氧胆酸(24)Fig.8 Multienzyme-catalyzed tandem synthesis of ursodeoxycholic acid(24)
尽管实现了UDCA(24)的酶法高效合成,但上述酶催化串联反应随着反应时间的延长,其体系中副产物7-oxo-LCA 会还原为CDCA,同时热处理灭活7α-HSDHEc又耗时繁琐,大大限制了该法进一步的工业化应用研究。对此,许建和课题组[69]又采用了固定化酶技术对UDCA 的酶法合成体系进行了优化。固定化酶技术是用物理法或化学法,将游离酶人工固定在特定载体上进行特有的催化作用,可回收并长时间使用的一种技术。固定化酶较游离酶具有稳定性高、易于控制、可反复使用、回收方便、成本低廉等优点,因此在生物工程、医学、能源开发等领域具有重要作用[70-71]。许建和课题组首先筛选了不同的固定化方法,最后将7α-HSDHEc和LDH、7β-HSDHRt-M1(突变体T189V/V207I)和GDH 分别在环氧化树脂ES-103 上进行了共固定化,并对固定化条件进行了优化。随后研究人员设计了一种流动式填充床反应器,以固定化酶LDH-7α-HSDH@ES-103 和7β-HSDH-GDH@ES-103 为催化剂,通过优化填充床反应器系统条件,实现了CDCA 到UDCA(24)的高效转化,其转化产率高达100%,时空产率约88.5 g/(L·d)(图8)[69]。这样一个将固定化技术与酶催化串联法结合大量合成UDCA(24)的实例为复杂天然产物的规模化制备提供了新的思路。
1.4 大环内酰胺类化合物
多环特特拉姆酸大环内酰胺(polycyclic tetramate macrolactams,PoTeMs)是一类具有特特拉姆酸结构单元及多环体系的大环内酰胺类化合物,具有多样的生物学活性[72]。斑鸠霉素(ikarugamycin,25)是第一个报道的5/6/5 三环PoTeMs 化合物, 1972 年从嗜色链霉菌(S.phaeochromogenes)中首次分离获得,具有抗菌、抗炎、抗肿瘤、抗原虫、抗溃疡等生物活性[73-74]。此外,斑鸠霉素(25)还可抑制巨噬细胞中氧化低密度脂蛋白诱导的摄取以及HIV-1 Nef 诱导的细胞表面CD4 受体蛋白的下调,被认为是一种网格蛋白依赖性的内吞抑制剂[75-76]。斑鸠霉素(25)因其多样的生物活性自发现以来就引起了有机合成化学家对其全合成的探索。然而,斑鸠霉素(25)结构的高度复杂性在很大程度上限制了高效化学合成方法的探索,目前化学全合成产率小于1%[72,77]。斑鸠霉素(25)的生物合成途径由三个酶负责催化:杂合聚酮合酶IkaA(iPKS/NRPS)负责形成特特拉姆酸(26)、氧化还原酶IkaB 负责5/6 双环体系的形成、乙醇脱氢酶IkaC 则负责斑鸠霉素(25)中内5 元环的形成[72,74]。此外,斑鸠霉素(25)中的环己烯基单元可能是通过自发的Diels-Alder 反应合成[72,74]。Greunke 等[72]通过采用酶催化串联一锅法在体外成功实现了斑鸠霉素(25)的全合成(图9)。研究人员首先成功表达了来源于Streptomycessp.Tü6239 的蛋白IkaA 和IkaB以及来源于Salinispora arenicolaCNS-205的同源蛋白IkaC。随后以乙酰辅酶A、丙二酸单酰辅酶A和L-鸟氨酸(L-ornithine)为底物,在反应体系中同时加入IkaA、IkaB 和IkaC 以及相关辅因子,实现了斑鸠霉素(25)的体外酶法全合成,产率约9%。此外,为了减少成本,研究人员将IkaA、IkaB和IkaC与合成乙酰辅酶A的乙酸激酶AckA和磷酸转乙酰酶Pta 以及与丙二酸单酰辅酶A 合成酶MatB 进行串联,一锅法在体外也成功实现了斑鸠霉素(25)的酶法全合成。仅利用3个酶,就可构筑斑鸠霉素(25)中15 个C—C 键、2 个C—N 键以及8个手性中心的形成,充分展示了酶催化的神奇之处,也充分体现了酶作为催化剂在复杂天然产物全合成中发挥的强大作用。
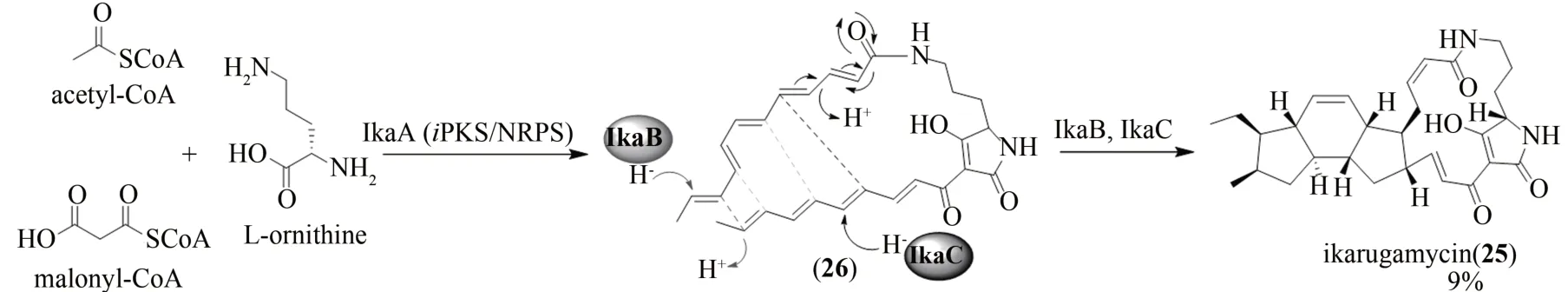
图9 斑鸠霉素(25)的生物合成途径Fig.9 Biosynthetic pathway of ikarugamycin(25)
1.5 UK-2A类似物
Fenpicoxamid(商品名Inatreq)是陶氏益农公司最新研发的一种新型吡啶酰胺类农用杀菌剂,其被真菌吸收后可在体内重新转化为UK-2A[78]。UK-2A 可特异性地抑制电子传递链泛醌Q1,阻断线粒体呼吸作用从而发挥抗真菌作用,且无交叉耐药性[79]。研究表明,UK-2A 的抗真菌活性主要来源于其C-3 位的3'-羟基-4'-甲氧基吡啶酸结构单元[80-81]。近日,Tan 等[82]通过多酶催化串联法成功实现了对UK-2A 药效官能团C-3-吡啶甲酸结构的定向改造。研究人员首先通过挖掘分析UK-2A 的生物合成基因簇并结合体外酶催化实验鉴定了7 个关键基因,阐明了UK-2A 的生物合成途径。其中,C-3-吡啶甲酸结构的合成是通过腺嘌呤苷酸结合蛋白UkaF 将3-羟基吡啶酸(3'-hydroxypicolinic acid)上载至肽基载体蛋白UkaG 上,然后经P-450 酶UkaL 和甲基转移酶UkaN 的修饰形成最后的C-3 位结构片段。同时,考察UkaF 的底物谱发现其具有宽泛的底物杂泛性,可识别13 种水杨酸(salicylic acids)及其衍生物。随后,研究人员通过多酶催化串联一锅法,以3-羟基吡啶酸或13 种水杨酸及其衍生物为底物,在体外成功合成了14 个新的脱酰基UK-2A(deacyl UK-2A,DAUK-2A)类似物[(27)~(40)],转化产率高达100%;首次实现了UK-2A 的C-3 位官能团的高效定向改造(图10)。
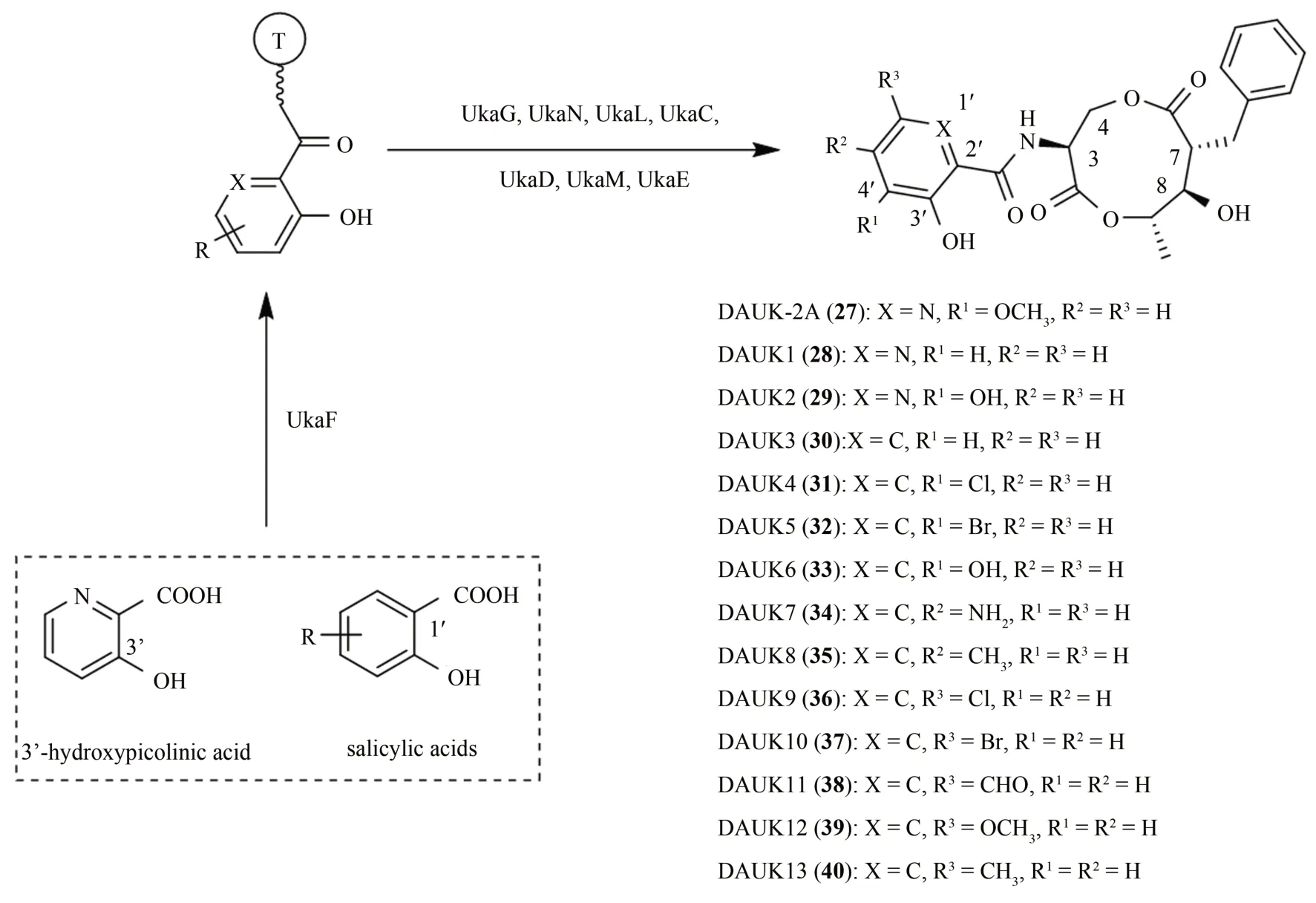
图10 多酶催化串联法合成UK-2A类似物Fig.10 Multienzyme-catalyzed tandem synthesis of UK-2A analogues
1.6 核苷类化合物
核苷类似物islatravir(原名MK-8591,41)是美国默克公司研发的一种HIV 逆转录酶移位抑制剂,目前正在进行临床试验研究,其非凡的功效和长效的作用有望在降低HIV治疗次数和暴露前预防感染中发挥作用[83-84]。Islatravir(41)主要通过化学合成获得,但化学合成存在步骤繁琐(需要12~18 步)且效率低等问题,主要在于难以控制2'-脱氧核糖的立体选择性[85-86]。鉴于此,Huffman等[87]在天然细菌核苷补救途径的启发下,采用逆向合成思路,通过定向进化改造五种天然酶使其作用于非天然底物,并通过采用多酶催化串联一锅法成功实现了islatravir(41)的经济高效合成(图11)。
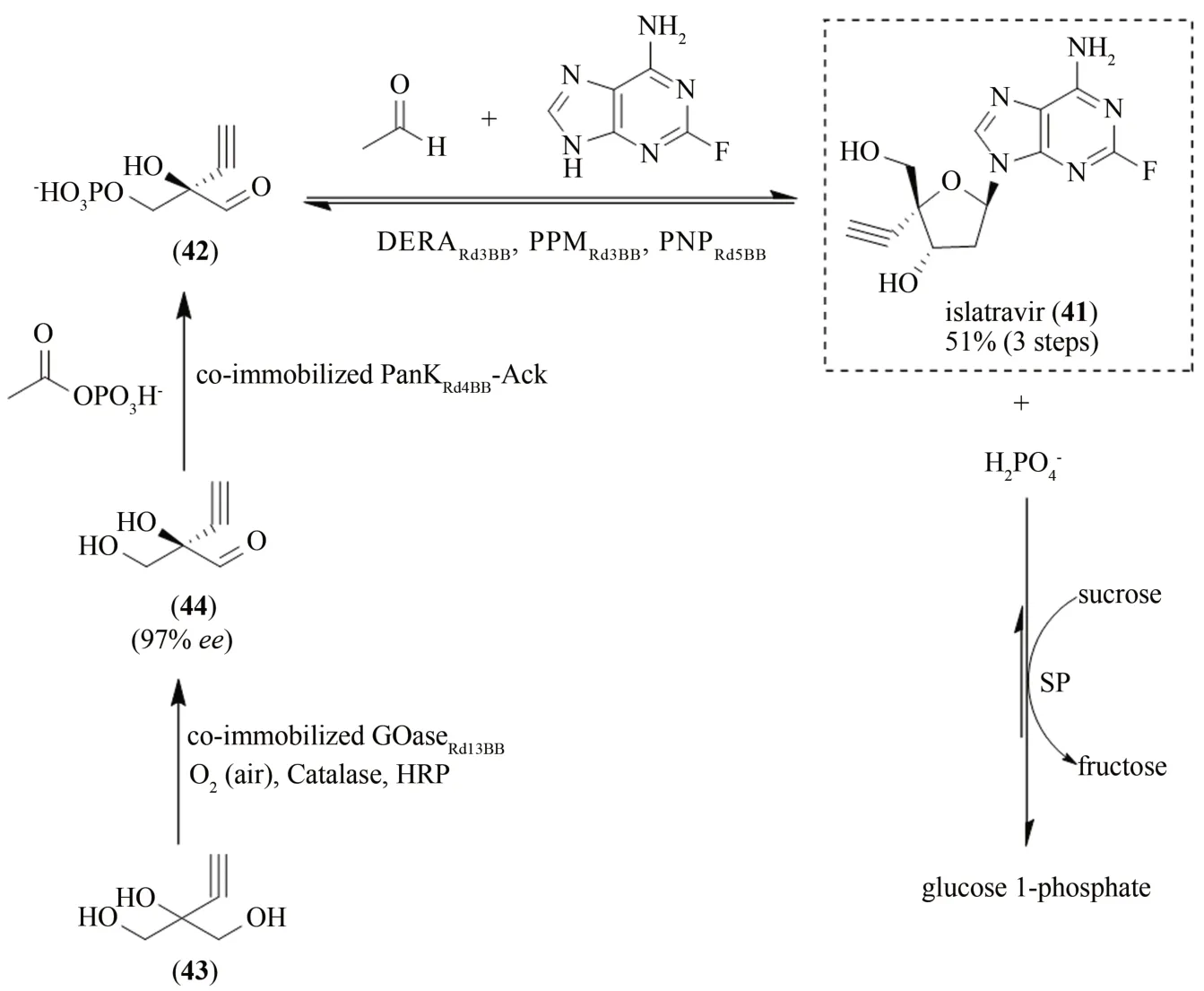
图11 多酶催化串联法合成islatravir(41)Fig.11 Multienzyme-catalyzed tandem synthesis of islatravir(41)
细菌核苷补救途径为脱氧核糖核苷提供了逆向合成方法,该过程主要使用三种酶降解嘌呤2'-脱氧核糖核苷:嘌呤核苷磷酸化酶(purine nucleoside phosphorylase,PNP)用磷酸替换核苷碱基得到脱氧核糖1-磷酸;随后磷酸戊糖变位酶(phosphopentomutase,PPM)将磷酸盐转移至5位;所得的5-磷酸糖在5-磷酸脱氧核糖醛缩酶(deoxyribose 5-phosphate aldolase,DERA)催化下将醛醇缩醛裂解为3-磷酸甘油醛和乙醛。当逆向合成时,该过程即可从简单的起始底物合成核苷[88-89]。基于此,研究人员充分利用了酶催化反应的可逆性,采用逆向合成策略进行islatravir(41)的合成。然而,islatravir(41)结构中具有的炔基取代基和氟取代基不易被天然酶所识别。对此,研究人员首先对核苷补救途径中的三种酶进行了探索研究。通过定向进化改造来源于E.coli的PNP 和PPM 以及来源于希瓦氏菌(Shewanella halifaxensis)的DERA,成功获得了对非天然底物具有高活性高耐盐的PNPRd5BB突变体和高活性的PPMRd3BB突变体,其活性较野生型酶分别提高了70 倍和350 倍;同时也获得了高选择性以及对乙醛高耐受的DERARd3BB突变体。由于PNP 和PPM 串联的反应被副产物磷酸盐所抑制,研究人员向反应体系中加入了蔗糖磷酸化酶(sucrose phosphorylase,SP)和蔗糖(sucrose),以消耗副产物无机磷酸盐,推动反应平衡。为了合成3-磷酸-2-炔基甘油醛(42),研究人员选择了对2-乙炔基甘油(43)先氧化再磷酸化的路线。通过对E. coli来源的泛酸激酶(pantothenate kinase,PanK)定向进化并将其与海栖热袍菌(Thermotoga maritima)来源的热稳定乙酸激酶(acetate kinase,AcK)配对,提高了PanK 的活性(较野生型提高100 倍)和稳定性,并实现了PanK 所需辅因子腺苷三磷酸的再生。在氧化还原酶的筛选中,研究人员鉴定了禾谷镰孢菌(Fusarium graminearum)来源的半乳糖氧化酶突变体(galactose oxidase,GOase)并对其进行定向进化,获得了具有高活性高选择性的GOaseRd13BB突变体。需要注意的是,GOase 催化的反应体系需要额外的过氧化氢酶和辣根过氧化物酶(horseradish peroxidase,HRP)。
实现了从2-乙炔基甘油(43)到islatravir(41)合成的每一步反应后,研究人员设计了一个3 步9 酶催化串联反应(图11)。但由于反应体系中引入的酶量较大,对终产物的过滤造成了困难,同时体系中的PanK 可直接催化初始底物2-乙炔基甘油(41)形成副产物。为此研究人员对PanK(配对Ack)和GOase 进行了酶的固定化操作,减少了水溶液中的酶量,并避免了初始底物的直接磷酸化。最终,通过优化酶催化串联反应平台,首次实现了islatravir(41)的规模化合成,其总产率高达51%,纯度高达95%[87]。Islatravir(41)的体外酶法高效全合成是酶催化串联法在复杂天然产物合成应用研究中的重大突破,其合成过程中涉及到的逆向合成思维以及定向进化和酶的固定化等策略,为设计合成其他复杂分子提供了新的思路和重要借鉴。
1.7 其他化合物
除上述介绍的重要实例外,采用体外酶催化串联一锅法合成的天然产物还有维生素B12的前体hydrogenobyrinic acid(45)[90]、聚酮化合物灰黄霉素(griseofulvin,46)[91]、杂萜类merochlorin A(47)和merochlorin B(48)以及napyradiomycin A1(49)和napyradiomycin B1 (50)[92-93]、植物抗毒素camalexin(51)[94]、吡咯并吲哚类生物碱毒扁豆碱(physostigmine,52)[95]、色氨酸衍生物indolmycin(53)[96]、硝基咪唑类抗生素azomycin(54)[97]、糖类化合物碳霉糖(TDP-L-mycarose,55)[98]、多肽类抗生素56[99]、以及免疫抑制剂lipid IVA(57)[100],其结构见图12。
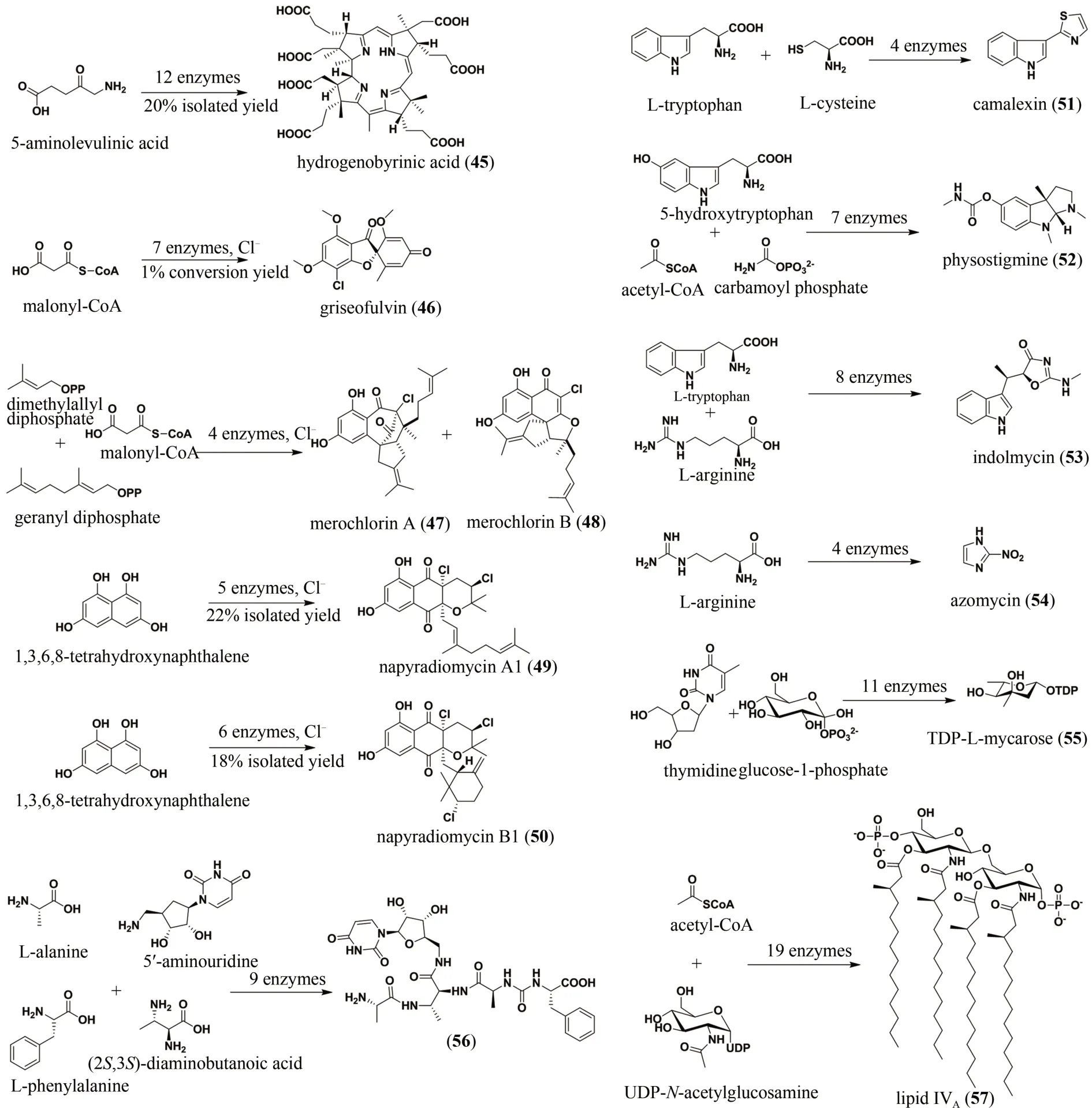
图12 酶催化串联法合成的一些其他化合物(45~57)Fig.12 Enzymatic tandem synthesis of other compounds 45~57
2 化学-酶催化串联法合成复杂天然产物
化学-酶催化串联就是将化学合成与酶催化合成进行串联,两者互补,各取所长,从而完成天然产物的合成。化学催化剂如有机金属催化剂具有宽泛的底物适用范围和催化活性,可以弥补某些酶对非天然底物没有催化活性以及在有机溶剂或高温下出现的活性丧失等不足;而酶催化剂能在更加简单温和的催化条件下将区域和立体选择性发挥到极致,可以解决化学催化剂在没有诱导剂或保护基团的辅助下难以实现极高手性选择性的问题。然而,化学-酶催化串联体系的实现较为困难,主要源于两类催化剂所需的催化环境不同;当生物酶和化学催化剂共存于同一反应体系时往往存在相互抑制的情况[101]。尽管如此,利用酶自身催化反应的高效性、高立体选择性、高区域选择性,以及利用化学催化剂具有的宽广的底物适用范围和催化活性,化学-酶催化串联在复杂天然产物尤其是高手性化合物的合成中发挥了重要作用[102-106]。近年来,无论是化学-单酶催化串联还是化学-多酶催化串联,在复杂天然产物的合成中均取得了巨大的应用进展[107-111]。关于化学-酶催化串联法在复杂天然产物合成中的应用研究进展,目前已有相关综述进行了较全面的总结[101-106,109]。本节以多杀菌素A(spinosyn A,58)[111]和肝素(heparin)[107]的化学-酶催化串联合成研究为例,介绍化学合成与多酶法合成相互结合在复杂天然产物合成中的重要性。
2.1 多杀菌素A 的合成研究
多杀菌素A(58)是刺糖多胞菌(Saccharopolyspora spinosa)产生的大环内酯类化合物,其作为生物杀虫剂多杀菌素的主要活性成分之一,在农业应用中占有重要地位[112]。多杀菌素A(58)分子结构复杂,以含有21 个碳所组成的独特的5/6/5/12-四环作为基本骨架,并通过氧糖苷键在C-9 位和C-17 位分别与三氧甲基鼠李糖和福乐氨糖连接,整个分子中含有17个手性中心。多杀菌素A(58)的化学全合成已经由Evans、Paquette、Roush 和Dai 四个课题组分别独立完成,但由于多杀菌素A(58)独特的四环结构以及分子中众多的手性中心,在合成过程中给他们带来了巨大挑战[103,113]。多杀菌素A(58)的生物合成途径已被解析(图13):其四环骨架结构(59)是由模块化Ⅰ型聚酮合酶SpnA-E 催化1 分子丙酰辅酶A、9 分子丙二酸单酰辅酶A 和1 分子甲基丙二酸单酰辅酶A 结合而成;随后在SpnJ 的催化下将15-OH 氧化为羰基得到化合物60,并在SpnM 催化下脱水生成多烯化合物61;化合物61 在SpnF 催化的[4+2]-环加成Diels-Alder 反应中形成化合物62,继而在鼠李糖转移酶SpnG 作用下进行糖基化,并由SpnL催化C-2 位和C-14 位相连形成化合物63;化合物63 在甲基转移酶SpnH、SpnI 和SpnK 的作用下对鼠李糖单元羟甲基化;最终在SpnP 的催化下引入福乐氨糖合成最终产物多杀菌素A(58)[114-116]。
基于化学催化可合成宽广的底物和酶催化反应具有很高的立体选择性和区域选择性,研究人员在2014 年采用化学-酶催化串联法成功实现了多杀菌素A(58)的全合成(图13)[111]。由于多杀菌素A 生物合成途径中化合物59 的结构只是推测而来,尚未分离鉴定,研究人员首先采用化学法合成了多烯大环内酯化合物59。化合物64 通过不对称乙基化和羟醛缩合反应等5步转化获得化合物65,随后发生Stille 偶联、Julia-Kocienski 烯基化以及Yamaguchi 大环内酯化,经7 步转化完成了酶催化前体59 的合成。随后研究人员以化合物59 为底物,通过在反应体系中加入多杀菌素A(58)生物合成途径相关的8 个酶以及优化体系中各酶的浓度,在体外一锅法实现了化合物66 的合成,其总转化产率约19.6%,意味着每一步酶催化反应的平均转化产率至少为81.6%。由于多杀菌素A(58)生物合成途径中最后一步SpnP 催化的糖基化反应在体外需要辅助蛋白作用才能发挥活性,而多杀菌素A(58)生物合成基因簇中并没有相关编码基因存在。因此,研究人员最后采用了化学方法,在Lewis 酸活化下对化合物66 引入氨基葡萄糖实现了多杀菌素A(58)的全合成。多杀菌素A(58)的化学-酶催化串联全合成工作充分体现了酶催化反应的高效性、专一性和简便性,同时也证实了化学合成与生物合成相结合的策略在复杂天然产物合成中的重要价值。
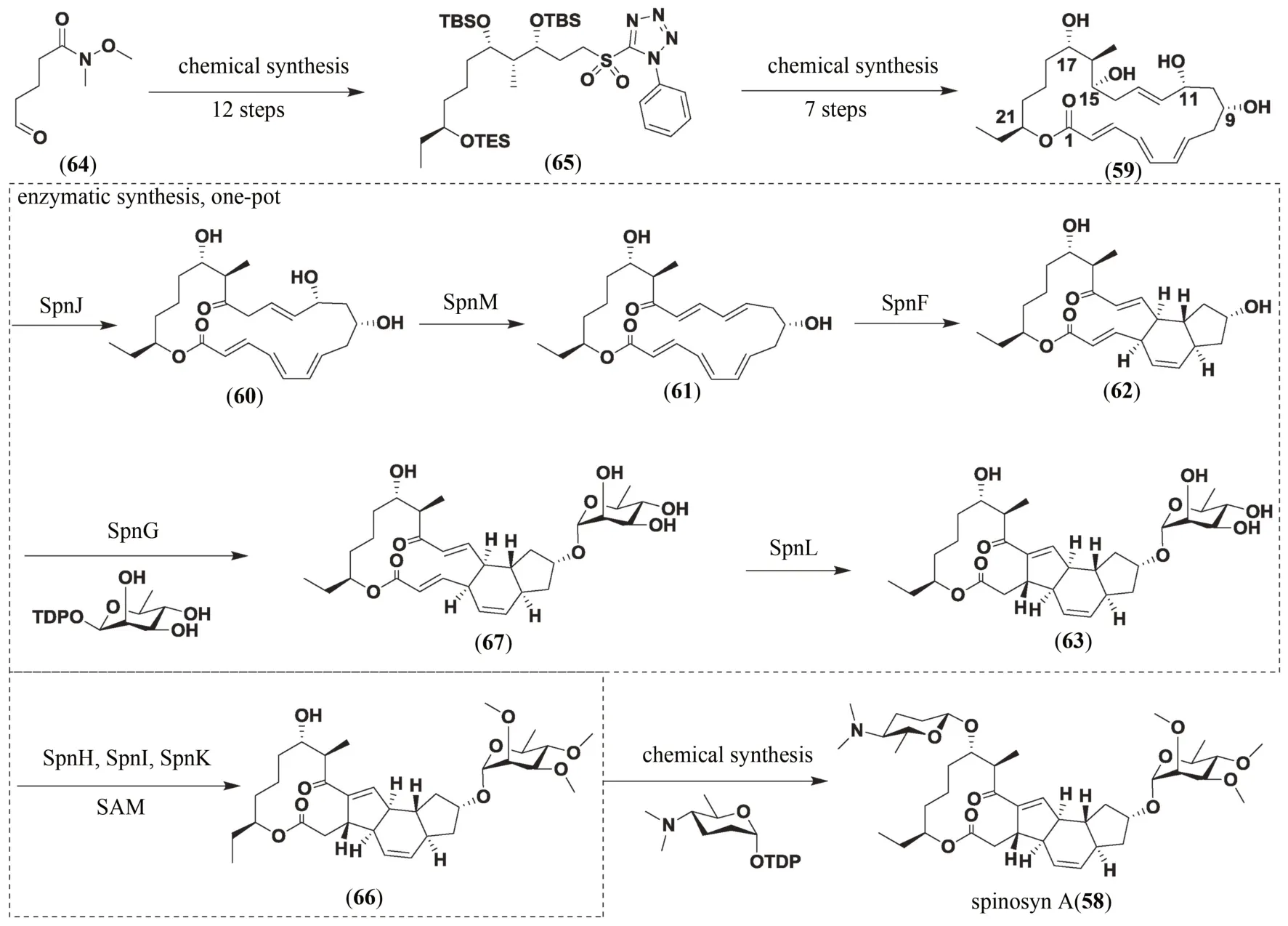
图13 化学-酶催化串联法合成多杀菌素A(58)Fig.13 Chemoenzymatic synthesis of spinosyn A(58)
2.2 肝素的合成研究
化学-酶催化串联合成的另一个经典的例子是2011 年报道的肝素的全合成[107]。肝素是临床上广泛使用的抗凝药物,是葡萄糖醛酸(glucuronic acid,GlcA)或艾杜糖醛酸(iduronic acid,IdoA)和葡萄糖胺(glucosamine,GlcN)通过1→4 糖苷键连接形成的二糖重复单元组成的糖胺聚糖。肝素大小不一、结构各异,第一个化学全合成的五糖抗凝药磺达肝素(商品名Arixtra)总共涉及50多步反应,且反应条件复杂、产率极低(小于0.1%),极大地限制了商业化应用[117-118]。肝素的体内生物合成首先是在N-乙酰葡萄糖胺转移酶(N-acetyl glucosaminyl transferase) 和肝素合酶(heparosan synthase-2,HS2)的作用下形成N-乙酰葡萄糖胺和葡萄糖醛酸交替连接的糖链骨架,随后在N-硫酸化酶(N-sulfotransferase,NST)、C5-异构酶(C5-epimerase,C5-epi)以及不同O-硫酸化酶(O-sulfotransferase,OST)的作用下对糖链骨架进行修饰,从而得到不同大小及结构的肝素。利用酶催化反应高效的立体专一性,Xu等[107]采用化学-酶催化串联法首次实现了两个超低分子量肝素[(68)和(69)]的合成(图14)。研究人员借助上述不同的糖基转移酶(来源于E. coliK5 的KfiA和来源于Pasteurella multocida的pmHS2)、硫酸化酶以及C5-异构酶,通过精细调控反应顺序,将天然葡萄糖胺、葡糖醛酸及硫酸根进行“组合”,并结合碱性(三乙胺,triethylamine)条件下的脱保护化学过程,只需10~12 步反应就高效合成了两个超低分子量肝素[(68)和(69)],其产率分别为45%和37%,且抗凝血活性与商品药磺达肝素相当。肝素化学-酶法全合成的实现是化学-酶催化串联法在复杂分子合成应用中的里程碑创举,其为抗凝血药物肝素的研发开辟了新的途径。

图14 化学-酶催化串联法合成超低分子量肝素(68)和(69)Fig.14 Chemoenzymatic synthesis of ultralow molecular weight heparins(68)and(69)
3 结语
实现天然产物的体外酶法全合成不仅可以拓展“酶”作为催化剂的应用潜力,也为复杂天然产物的制备提供了新的思路。体外酶催化合成具有高效性、高选择性、反应条件温和等优点,在复杂天然产物的合成中发挥了独特的优势。然而,体外酶法全合成的发展仍面临诸多挑战。首先,尽管复杂天然产物的生物合成研究近年来取得了很大进展,但与化学合成相比,人们对于由生物大分子负责的生物合成机制的理解仍显不足。天然产物的体外酶催化全合成需要在其生物合成途径了解的基础上进行,这样才能保证酶催化反应中初始底物的真实性以及酶催化串联反应的有效性。然而一方面天然产物结构多样,另一方面相似化合物的生物合成途径也是共性与差异共存,因此人们还需继续努力挖掘生物合成基因簇和阐明更多的生物合成途径。相信随着测序技术、生物信息学以及基因编辑、结构生物学、遗传转化等生物技术的迅速发展,越来越多复杂天然产物的生物合成途径将得到解析,从而为复杂天然产物的体外酶催化全合成奠定基础。其次,体外酶催化反应的实现需要生物合成途径中所有参与反应的酶在体外能够稳定获得且有活性,但目前仍有很多目标蛋白无法通过常规的生物技术获得、或蛋白不稳定、或活性很低,极大限制了体外酶催化全合成的应用。另外,复杂天然产物生物合成途径中参与反应的酶的功能多种多样、酶学性质不同,在体外将它们串联在一起时容易出现蛋白互不兼容、反应条件互不合适、中间体产物及辅因子容易抑制一些酶的活性等问题,很大程度上影响了酶催化全合成的效率。对此,可通过优化表达系统、筛选同源基因、构建融合蛋白等来获得目标蛋白;通过优化反应条件、固定化、定向进化等技术提高酶的稳定性和兼容性。此外,也可通过分步串联策略或多酶催化串联流动体系提高蛋白稳定性、减少中间体产物及辅因子对酶活性的抑制,从而最大程度地提高体外酶催化串联体系的合成效率。在体外酶催化途径的设计中,应该综合考虑化学合成和酶催化的优势,不拘泥于全酶串联,在一些特殊步骤中引入化学合成也将大大提高整体合成效率。综上所述,天然产物的体外酶催化全合成研究近年来取得了较大进展,相信随着合成生物学理念的进一步完善,蛋白质工程和酶工程技术的发展,体外酶催化全合成将成为复杂天然产物合成的重要手段之一。
猜你喜欢
故事作文·低年级(2023年11期)2023-12-05 06:39:56
故事作文·低年级(2023年12期)2023-03-24 14:16:52
云南化工(2021年6期)2021-12-21 07:30:56
云南化工(2020年11期)2021-01-14 00:50:48
应用化工(2020年9期)2020-09-29 08:55:16
科学(2020年2期)2020-08-24 07:57:00
中国环境监察(2016年7期)2016-10-23 05:36:30
中国现当代社会文化访谈录(2016年0期)2016-09-26 08:46:23
生物技术通报(2015年1期)2015-04-10 16:15:19
中南民族大学学报(自然科学版)(2014年4期)2014-08-06 05:49:24
