
合成生物学在感染性疾病防治中的应用
2020-08-07 05:52:52蒲璐黄亚佳杨帅金帆
合成生物学 2020年2期
蒲璐,黄亚佳,杨帅,金帆
(中国科学院深圳先进技术研究院,深圳合成生物学创新研究院,中国科学院定量工程生物学重点研究室,广东 深圳 518055)
合成生物学是生物科学在21 世纪出现的一个分支学科。在近20 年的时间里,合成生物学已经发展成为一门通过结合工程学和生物学原理,以系统合理的方法对标准化的生物元件进行重组、创造和设计,来获取具有可预测性和应用性的功能型生物装置、系统或生物体的科学[1]。随着对可用基因模块的不断开发,以及对生物分子模块的进一步了解,并结合数学建模,合成生物学家们可以借鉴基因和基因组的基本要素及其组合原理,来设计、合成和重建已存在的基因元件或构建新的基因零件来重塑生命系统[2-4]。
合成基因回路中包含了基本的信号响应调控元件,这些元件可以受特定的外源刺激或内源性代谢产物的激活,对后续基因表达进行调控[5]。这些可以控制基因的“开关”通常是触发诱导型蛋白质和DNA 之间[6-7]或是适配体和转录开启之间[8-9]的相互作用,反过来说,信号响应调控元件与调控因子的相互作用通过调控转录翻译,响应外源性或内源性的输入信号。基因开关的标准化设计改善了不同功能基因在同一时空表达的兼容性[10],使得复杂基因网络的构建成为可能,如多触发信号输入和时序控制[11-12]、共同控制[13-14]和反馈控制[15]回路元件等,同时也使得可预测功能性的复杂蛋白能够实现高精度原位的动态表达。
合成生物学的理念和方法被运用到生物医学中,为现代医学注入了新的思路和策略。合成生物学疗法的基本原理,如图1 所示,是通过将人工构建的生物分子或“编程化”的有机体等人工合成生物系统转入宿主内,重建内稳态,调节疾病引起的系统紊乱或功能异常等[16]。合成生物学已在感染性疾病、代谢性疾病、神经退行性疾病和癌症等的诊断治疗和预防上显示出巨大的潜力[17]。本文作者着重介绍合成生物学在感染性疾病诊疗和预防中的研究进展。
1 感染性疾病概述及其治疗瓶颈
一般将感染性疾病[18]定义为由病原微生物引起的机体疾病,常见的如炎症或器官功能障碍等。感染性病原体的种类包括细菌、病毒、真菌等。其中细菌感染性疾病有鼠疫、百日咳、猩红热以及细菌性败血症等;病毒感染性的疾病种类较多,最常见的是流行病毒引起的感冒,另外还有病毒性肝炎、麻疹、带状疱疹等;真菌感染导致的疾病,常见的比如脚气病,就是皮肤癣菌引起的足部皮肤真菌感染[19]。病原菌更容易侵袭免疫力低下的人群,引起各种感染性疾病。
针对不同的病原体引起的感染,有相对应的诊断和治疗方案。利用抗生素、抗毒素等直接消灭病原体或消除毒素的药物治疗是感染性疾病的基本治疗手段。然而,随着抗病原体药物的广泛长期使用,病原体的耐药性日渐增强,远快于人们研制新抗病原体药物的速度。某些地区抗生素的滥用有时反而使感染概率增加或感染加重,特别对一些自身有免疫缺陷病的患者,用抗病原体药物已经难以控制其体内的病原体感染[20]。
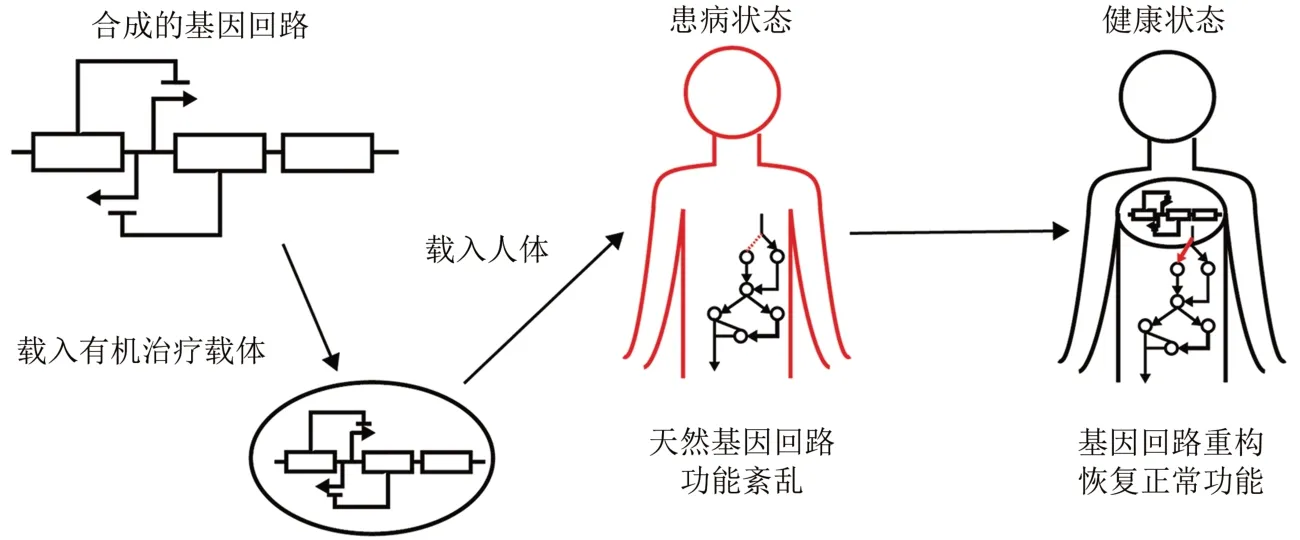
图1 合成生物学治疗原理[16]Fig.1 Schematic for synthetic biology therapy[16]
2 合成生物学对抗感染性疾病的优势
为了解决病原体造成的人类健康问题,研究者们开始尝试新的方法,合成生物学的理念也因此被应用到感染性疾病的治疗和预防中。在过去十几年里,针对感染性疾病防治的工程化合成生物学医学元件逐步迈向临床化,其中一部分现已经处于从理论、体外试验,动物实验到临床试验的过渡阶段。
相比于传统依靠化学合成获取抗病原体药物,合成生物学通过对现有生物系统进行改造,实现微生物自我繁殖,不需要建立大型化工厂,成为更加高效、清洁、符合市场期待的新兴技术。进入21世纪后,新抗病原体药物的研发进入瓶颈期,以“造物”为特色的合成生物学的出现正好满足了人们的期望。不同于大多数的广谱抗病原体药物,合成生物学疗法通常针对特定的目标病原体,并且可以根据病人的实际情况给出定制性的方案。此外,随着计算机科学与合成生物学相结合,通过算法、模型、机器学习等手段来设计基因回路,预测合成效果,模拟代谢路径,记录过程与结果,优化体系,使得合成效率和效果得到不断的提升,大大缩短了开发新药的时间,这些都是传统治疗手段无法比拟的。
如今,合成生物学已在感染性疾病的领域做出了初步尝试,逐渐改变了对病原体的诊断和治疗方式;在细菌感染性疾病中显示出了理想的治疗效果和巨大的潜力;为病毒感染性疾病致病机理的研究提供了新的思路;并参与到了感染性疾病预防的研究中。
3 合成生物学在感染性疾病诊断中的应用
合成生物学在疾病诊断方面已展现出巨大的优势。合成生物学家通过人工设计基因回路改造人体自身细胞、细菌、病毒等生命体,这些人工构建的生命体能够感知疾病特异性信号或人工信号、特异性靶向异常细胞和病灶区域,表达报告分子,达到疾病诊断的目的。有的人工改造生命体同时具备诊断和治疗的功能,会在检测到目标信号的同时,释放治疗药物。
聚合酶链式反应(polymerase chain reaction,PCR)技术是目前核酸检测的主流方法。而近年来,基于规律成簇的间隔短回文重复(clustered regularly interspaced short palindromic repeats,CRISPR)技术的检测手段因其快速、准确、经济、操作简便、对专业设备和基础设施要求低等特性正快速发展起来。
2014年,Collins等[21]首次将CRISPR/Cas技术引用核酸分子诊断,开发了一种低成本的能区分出寨卡病毒(Zika Virus,ZIKV)亚型的纸基传感器。这种检测方法首先是从样本中提取RNA 靶标并进行扩增,然后将扩增产物滴加到含有冻干细胞组分和生物蛋白的纸片上,激活冻干组分发生生化反应,纸片会发生肉眼可见的颜色变化,从而指示ZIKV 的存在。在此检测平台的帮助下,可以辨别临床症状相似的ZIKV 和登革热病毒(Dengue Virus,DENV),正确诊断出患者所感染的病毒类型仅需3h。当检测到ZIKV 时,将样品与冻干的CRISPR/Cas9 混合,利用CRISPR/Cas9 对PAM 区域优异的序列识别能力,还可以区分不同亚型的ZIKV,实现现场快速准确的毒株特异性诊断。
2016 年,张锋等[22]鉴别出可以被用来切割细菌RNA 序列的CRISPR/Cas13a 系统,深入研究发现该系统不同于靶向DNA 的CRISPR/Cas9 或CRSPR/Cas12a系统,Cas13a切割完靶标RNA后依然保持活性,会继续切割非靶标RNA,这种非特异性切割后来被称为附属切割(collateral cleavage)[23]。在此基础上,张锋课题组与Collins课题组[23]合作将CRISPR/Cas13a 系统与重组酶聚合酶等温扩增 (recombinase ploymerase amplification,RPA)技术结合,使样本中极微量的核酸能够在37℃、0.5h 内实现数量级的扩增,同时加入了一个单链RNA 荧光报告探针,组成新的诊断工具“SHERLOCK”(specific highsensitivity enzymatic reporter unlocking),用来检测ZIKV、DENV 或其他病原体等。如图2 所示,SHERLOCK 的工作流程是:靶标RNA 经RPA 和T7 RNA 聚合酶转录扩增后,激活Cas13a的附属切割活性,切割底物产生荧光信号,经检测灵敏度已达到5×10-14mol/L。2018 年,张锋团队[24]再次升级了SHERLOCK 技术,推出SHERLOCK v2,新版本的检测系统中引入了3种来自不同种属细菌的Cas13 蛋白和Cas12a 蛋白(LwaCas13a 、PsmCas13b、CcaCas13b 和AsCas12a),能在同一样品中检测出多种病毒感染,如能同时检出ZIKA和DENV,从只检测1 个靶标RNA 进化到同时检测4 个靶标RNA,发出4 种不同颜色的荧光信号;并利用CRISPR type-Ⅲ中的Csm6酶,设计CRISPR系统使得Cas13 剪切后的序列是Csm6 酶的靶向序列,放大其测试信号,进一步增强了该方法的灵敏度,定量测量浓度最终低至2×10-18mol/L,敏感度提高了100倍。同时,该团队还开发了一种玻璃纤维试纸条,SHERLOCK v2 组分可以在纸上冻干再水化,90min就可以检测到浓度2×10-8mol/L以上的ZIKA 和DENV 的单链RNA。2019 年,Collins等[25]开发了一种可批量应用的、可编程的CRISPR 响应智能材料体系,可用于便携、精确、快速、定量且时空可控地检测危险病毒病原体,区分病原菌、人类DNA 的基因型。张锋等[26]将Cas13 的抗病毒活性与其诊断能力结合起来,建立了一个强大和快速可编程的诊断和抗病毒系统,命名为CARVER(Cas13-assisted restriction of viral expression and read out),靶向病人细胞中淋巴细胞性脉络丛脑膜炎病毒(LCMV)、甲型流感病毒(IAV)和疱疹性口炎病毒(VSV)的单链RNA,使细胞中病毒RNA 的水平降低了约30%,流感病毒的感染力降低了约45%,CARVER 技术使CRISPR/Cas13 技术迈向靶向治疗以及抗病毒药物开发的新领域。
2018 年Doudna 等[27]发现当Cas12a 蛋白在剪切靶向的dsDNA 后能高效地切割非特异性单链DNA(ssDNA),基于此开发了DETECTR(DNA endonuclease targeted CRISPR trans reporter)技术。与SHERLOCK 类似,DETECTR 将RPA 与Cas12a相结合,扩增产物激活Cas12a 的附属切割活性,切割底物产生荧光信号(图2)。DETECTR 技术可以准确地检测出是否有高危型人乳头瘤病毒(human papilloma virus, HPV) 感染: HPV16(100%)、HPV18(92%),在单核苷酸多态性分析、癌症筛查、细菌和病毒感染检测及耐药性筛查等方面有广泛的应用潜力。
中国科学院上海植物生理生态研究所赵国屏院士团队王金博士[28]也发现了Cas12a 对于靶标ssDNA 和非靶标ssDNA 的切割特性,并基于此原理开发了 HOLMES (an one-hour low-cost multipurpose highly efficient system)[29]技术。HOLMES 技术可以准确检测出诸如日本脑炎病毒(Japanese encephalitis virus, JEV)和伪狂犬病病毒(Pseudo Rabies Virus,PRV)等。
4 合成生物学在细菌感染性疾病治疗中的应用
合成生物学在细菌感染性疾病治疗中的应用,主要集中对细菌生物被膜的治疗中。
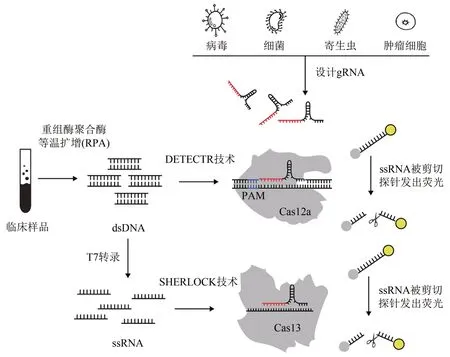
图2 基于CRISPR/Cas系统的SHERLOCK[23]和DETECTR[27]检测技术原理示意图Fig.2 Schematic for molecular diagnostic testing using CRISPR/Cas based technologies SHERLOCK[23]and DETECTR[27]
4.1 细菌感染性疾病与生物被膜
细菌感染是致病菌或条件致病菌侵入宿主体内,产生毒素和其他代谢产物所引起的局部或全身性感染。按病情的缓急程度,可以将细菌感染性疾病分为急性感染和慢性感染。实际上,急性感染和慢性感染是细菌感染宿主两个不同过程的结果,这两种类型的感染涉及细菌内部不同的分子机制。发生急性感染时,细菌在宿主体内处于浮游状态,快速的生长及传播;而发生慢性感染时,细菌会选择在宿主体内定殖,形成生物被膜,这个过程相对慢一点,但会造成长期持续性的感染。抗生素问世后,在相当长的一段时间里,细菌感染都受到了控制,尤其是急性细菌感染,抗生素治疗往往能收到良好的治疗效果。然而近几十年来,抗生素用量不断加大,疗效却难以令人满意,抗生素不得不更新换代,却依然赶不上耐药菌株出现的速度。随着现代医学的发展,高分子材料在医疗领域应用广泛,留置尿管、中心静脉插管、气管插管等操作引起的导管相关感染[30](catheter related infection,CRI)越来越常见,已经成为院内感染的主要原因,占医院感染的80%以上。造成CRI的重要因素之一是导管在体内留置时间过长,细菌在其表面黏附定殖并形成生物被膜。细菌生物被膜也是造成长期慢性细菌感染和细菌出现抗生素耐药性的重要原因之一[31]。
细菌生物被膜[32]是细菌黏附于接触表面,分泌多糖基质、纤维蛋白、脂类等,将其自身包裹在其中而形成的大量细菌聚集体。如图3所示,生物被膜的形成[33]通常有以下几个步骤:初始阶段,少量细菌可逆地黏附在接触表面;然后细菌开始繁殖,通过菌体表面黏附性物质将彼此黏结在一起,发展形成微菌落;以微菌落为基本单位,逐渐形成成熟的生物被膜;生物被膜中的细菌游离到环境中去,寻找新的定殖表面。
细菌生物被膜是细菌为适应环境,帮助自身生存的一种生命形式。细菌生物被膜相关的细菌慢性感染难以根治的原因有以下几点:①细菌在宿主体内形成的生物被膜使得细菌具有很强的耐药性[34],生物被膜内的细菌几乎对所有的抗生素都不敏感,即使依靠高于正常剂量成百倍的药物也不易彻底治愈;②生物被膜内的细菌能抵抗和干扰宿主的防卫机制,具有极强抗免疫系统的能力,造成持续性感染;③生物被膜是细菌感染的病灶,从生物被膜中游离出来的细菌除了能进一步扩大感染,也具有更强的毒性[35]。针对细菌慢性感染的治疗,更多的是针对细菌生物被膜的治疗,合成生物学家们通过改造噬菌体或工程菌,使其可以瓦解生物被膜,杀死具有强耐药性的细菌,或者阻断细菌对抗生素抵抗机制,削弱这些细菌的耐药性,起到辅助治疗的作用。
4.2 工程噬菌体在生物被膜治疗中的应用
噬菌体是侵袭细菌的病毒,有严格的宿主特异性。鉴于噬菌体是自然存在的细菌的“猎杀者”,噬菌体成为治疗细菌感染的一种选择。
在早期具有代表性的一例工程噬菌体研究中,Lu和Collins[36]通过改造本身就是大肠杆菌宿敌的T7 噬菌体,使其能合成特定的酶来瓦解细菌生物被膜,进而杀死被保护在生物被膜中的细菌。他们成功构建了能够持续表达DspB 蛋白的裂解性噬菌体T7-DspB,DspB 蛋白是能够水解β-1,6-N-乙酰基-α-葡萄酰胺的酶,而被水解的物质是生物被膜胞外聚合物重要的组成成分,即DspB 能够破坏生物被膜的外部结构。改造后的工程噬菌体T7-DspB 在消除生物被膜的过程中采取双管齐下的攻击策略,如图4 所示,在T7-DspB 噬菌体开始侵袭生物被膜时,不断地增殖和表达DspB,随着宿主菌被裂解,新生代的T7-DspB 噬菌体和DspB 也被释放到生物被膜中,继续感染其他细菌,同时降解生物被膜。在T7-DspB 噬菌体感染生物被膜体外实验测试中,生物被膜中的细菌数减少量达到99.997%,杀菌效果是使用未改造的噬菌体感染生物被膜的约4.5倍。
Lu 课题组[37]于2019 年开发了一种高通量的筛选方法,能够迅速扩大噬菌体的宿主范围,减缓细菌耐药性的产生。他们通过自然进化和结构建模,发现了T3 噬菌体尾丝蛋白中的宿主范围决定区域(host-range-determining regions,HRDRs),接着通过位点定向突变对这些区域进行高通量的筛选。受到抗体特异性工程的启发,这种方法探索出尾丝蛋白深层功能多样性的同时,最小化了对整体尾丝蛋白结构的影响,从而获得了合成的“噬菌体”。研究发现,高通量突变HRDRs 筛选出的噬菌体具有广泛的宿主适用性,这些噬菌体还通过减缓细菌耐药性的出现,在体外试验中表现出长时间对细菌生长的抑制性,并在小鼠模型中展现出相同的抑菌效果。
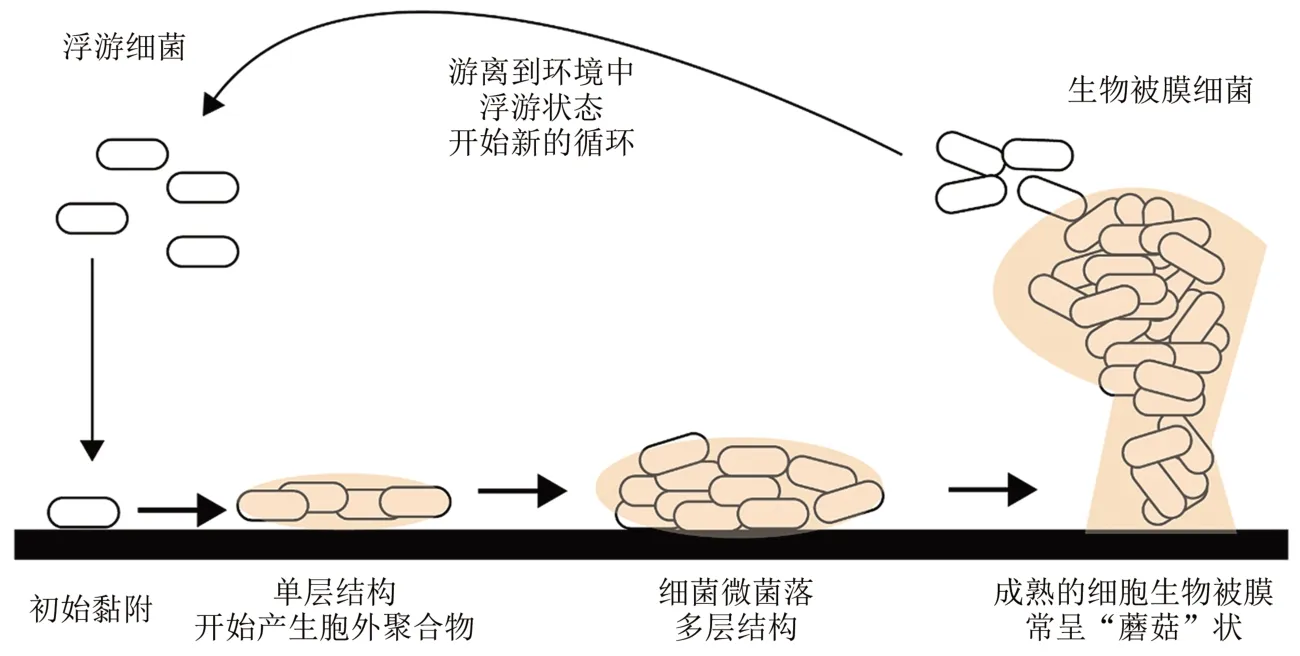
图3 生物被膜形成过程示意图[33]Fig.3 Schematic of biofilm formation[33]
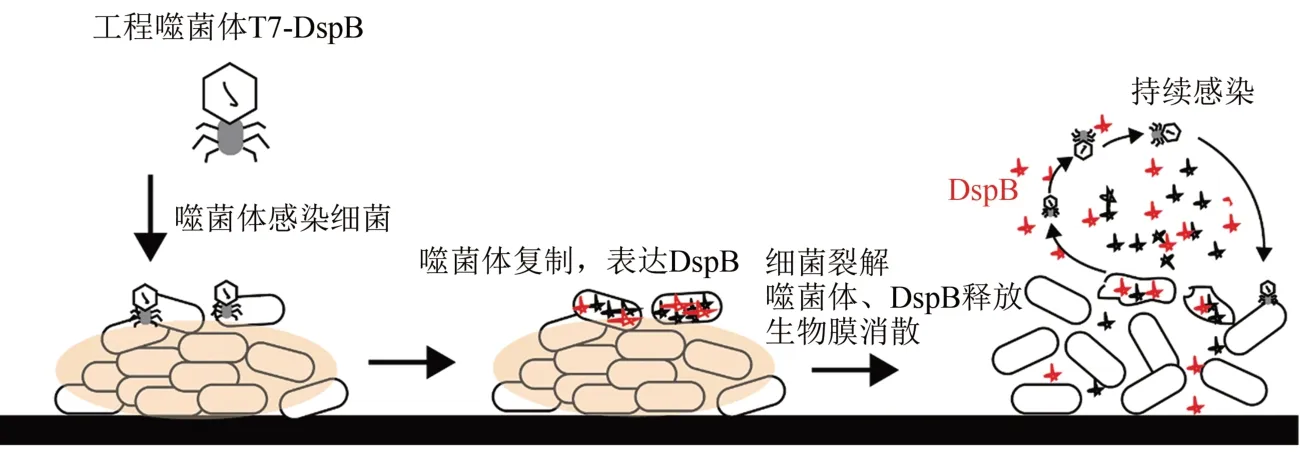
图4 利用工程噬菌体T7-DspB消除生物被膜的策略示意图[36]Fig.4 Schematic for dispersing biofilms with engineered bacteriophage T7-DspB[36]
虽然科学家们已经在利用工程噬菌体防治生物被膜中取得一系列进展,如工程噬菌体治疗单一或多菌株生物被膜[38-42]后,生物被膜明显消散,然而由于生物被膜的复杂结构,体外体内环境的差异性等原因,完全瓦解病人体内的生物被膜依然是亟待解决的科学问题。工程噬菌体和其他抗菌药物的联合使用,是治疗细菌感染的另一种可选方案。
Ryan 等[43]测试了联合使用T4 噬菌体和头孢噻肟消除大肠杆菌菌株ATCC11303 形成的生物被膜的效果。实验结果显示,使用104PFU/mL 和107PFU/mL的噬菌体可将头孢噻肟治疗大肠杆菌生物被膜的最小生物被膜清除浓度分别再降低2倍和8 倍,噬菌体和抗生素的联合使用优于单独使用抗生素消除生物被膜的效果。比起单独使用抗生素治疗,噬菌体和抗生素联合使用的效果在其他菌株,如金色葡萄球菌[44]和克雷白氏肺炎杆菌[45]等菌株的生物被膜消除中,也有明显提升的效果。通过合成生物学改造后的噬菌体联合抗生素使用,在一些情况下会展示出更好的结果。诸如环丙沙星等抗生素会破坏细菌的DNA,给细胞带来氧化压力,致使细菌启动SOS 响应的DNA 修复系统[46]。Lu 和Collins[47]通过改造M13 噬菌体,使其能表达LexA3,而LexA3 可以抑制细菌SOS 应激机制的开启,如图5 所示。工程噬菌体M13-LexA3显著增强了三类抗生素的杀菌效果,他们分别是喹诺酮类、β-内酰胺类和氨基糖苷类抗生素。在体外试验中,工程噬菌体M13-LexA3 和喹诺酮的联合使用杀死耐药菌的效率比单独使用喹诺酮时的杀菌效率,提升了约5000倍。在动物实验中,单独使用氧氟沙星治疗被大肠杆菌感染的小鼠,小鼠存活率仅为20%,而由工程噬菌体M13-LexA3和氧氟沙星联合治疗的感染小鼠,存活率达到了80%。
我国上海噬菌体与耐药研究所成立于2017年[48],2018 年初启动了国内首个伦理审批的噬菌体治疗临床试验,并于同年8月成功治愈了一例超级细菌感染者。患者是一位膀胱肿瘤术后复杂性、反复性尿路感染者,2014 年因全尿路内滋生了多重耐药的肺炎克雷伯菌,在左、右肾盂和膀胱3处中建立了感染。这几处的感染既有联系,又有差异,治疗难度相当大。治疗团队利用改造后可以识别并杀死肺炎克雷伯菌的噬菌体,确定了最终的噬菌体-抗生素联合治疗方案:肾盂造瘘后通过造瘘管进行噬菌体冲洗结合抗生素静滴治疗1 周,而后停用抗生素并继续噬菌体治疗1周,之后全面停药,连续观察1个月。最终,患者泌尿系统的肺炎克雷伯菌被彻底杀灭,尿路刺激症状显著改善,明显提高了患者的生活质量。
4.3 工程细菌在生物被膜治疗中的应用
随着合成生物学的发展,我们能够构建出具有不同功能的工程细菌,并且将这些“智能”细菌应用到临床治疗中[49]。细菌感染是临床上致病性和死亡率的重要因素之一,结合不同菌株间的竞争和同种菌株之间的联系等机制,科学家们构建出了可以自动检测病原菌、并杀死病原菌的工程菌株。
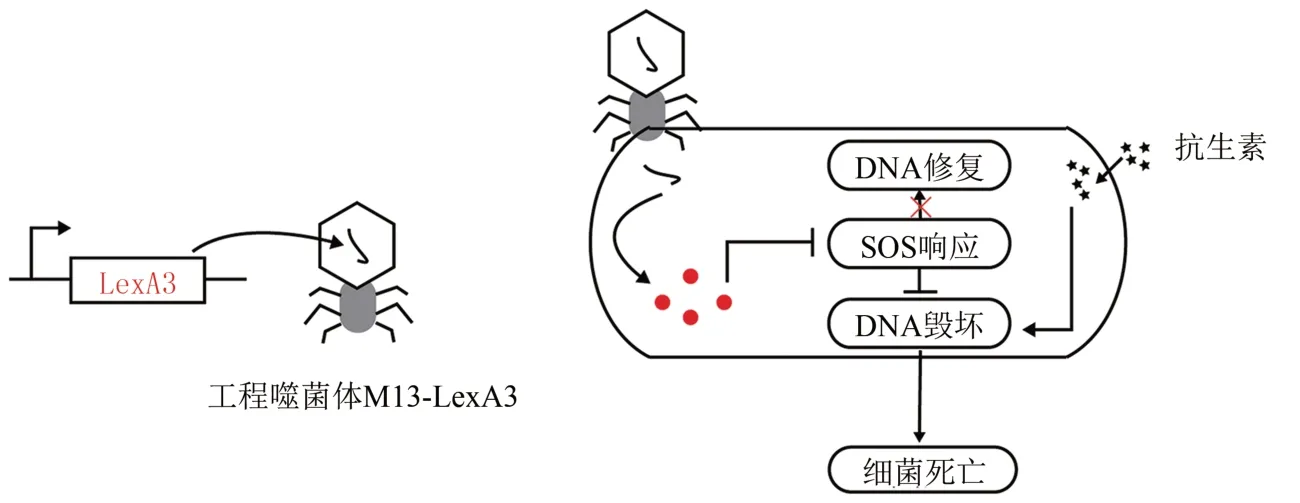
图5 利用工程噬菌体M13-LexA3增强抗生素对大肠肝菌EMG3菌株的杀菌效率示意图[47]Fig.5 Schematic for enhancing the efficacy of antibiotics treatment of E.coli through engineering with the phage M13-LexA3[47]
Chang等[50]在实验菌株大肠杆菌Top10中,设计并合成了一套包含群体感知、杀菌和裂解装置的系统,使得大肠杆菌能够感知并杀死临床分离的致病菌铜绿假单胞菌菌株Ln7。感知装置部分基于铜绿假单胞菌I型群体感知机制设计,如图6所示,启动子tetR持续表达转录因子LasR,LasR 会与铜绿假单胞菌释放的群体感知信号分子酰基高丝氨酸内酯(AHLs)3OC12HSL 结合,导致启动子luxR被LasR-3OC12HSL 复合物[51]结合开启,在感知装置中作诱导启动子;luxR启动子进一步激活系统中的杀菌和裂解装置部分,这两部分分别产生绿脓菌素S5 和大肠杆菌的裂解蛋白E7;当裂解蛋白浓度达到阈值时,大肠杆菌细胞膜破裂,释放出累积的绿脓菌素S5。绿脓菌素S5 是可溶蛋白,会分散到目标致病菌中,可以破坏目标菌株结构的完整性,从而杀死细菌。实验结果显示,工程大肠菌株可以杀死溶液中90%的活细菌,并且可以抑制铜绿假单胞菌生物被膜的形成,抑制率接近90%。
在对抗生物被膜的研究中,本课题组[52]利用光遗传学技术优化了的含有蓝光感受器LOV结构域蛋白的光遗传学工具质粒pDawn[53],并将之导入生物被膜模式细菌——铜绿假单胞菌的基因组,使铜绿假单胞菌在光照条件下适量表达功能性蛋白PA2133。PA2133是在铜绿假单胞菌基因组上的一段基因,它含有EAL结构域,能表达特定的磷酸二酯酶,从而水解环二鸟苷酸(cyclic diguanylate,c-di-GMP)。cdi-GMP[54]是细菌内广泛存在的一类重要的第二信使分子,调控细菌生物被膜的形成。这样构建出来的工程菌在光照条件下难以形成生物被膜,而在黑暗条件下形成的生物被膜通过光照,也会消散85%以上。
随着对人体体内微生物群的深入研究,益生菌也被选用为工程菌的基础菌株[55],为控制感染提供新的思路。Chang 等[56]对上述杀菌体系进行了升级,选用了益生菌菌株大肠杆菌Nissle 1917作为基础细菌进行改造,如图7所示,在原装置基础上添加了能够分解生物被膜胞外聚合物重要组成部分的酶DspB,并将承载合成基因回路的质粒,由抗生素识别标记改为了营养缺陷型识别标记,解决了水平基因转移带来的风险问题。这一工程益生菌在两种动物模型,秀丽隐杆线虫和小鼠模型中,对铜绿假单胞菌引起宿主的肠道感染都表现出了良好的预防和治疗作用。
4.4 CRISPR技术在细菌感染性疾病治疗中的应用
CRISPR 系统是细菌在对抗入侵病毒和外来质粒DNA 中演化出的防御机制,现在通过合成生物学技术的重新编程,可以用细菌自己的武器来对抗细菌病原体。研究人员成功地将CRISPR 干扰(CRISPR interference,CRISPRi)技术用于结核分枝杆菌感染模型,对目标基因进行有效的转录抑制,其作用机制涉及抑制细菌生长,改变其对小分子抑制剂的敏感性及破坏正常细胞形态。这是治疗多重耐药细菌的有利工具,尽管结核病早已可以治愈,但它仍是低收入发展中国家十大死因之一,每年造成全球约170万人死亡[57-59]。
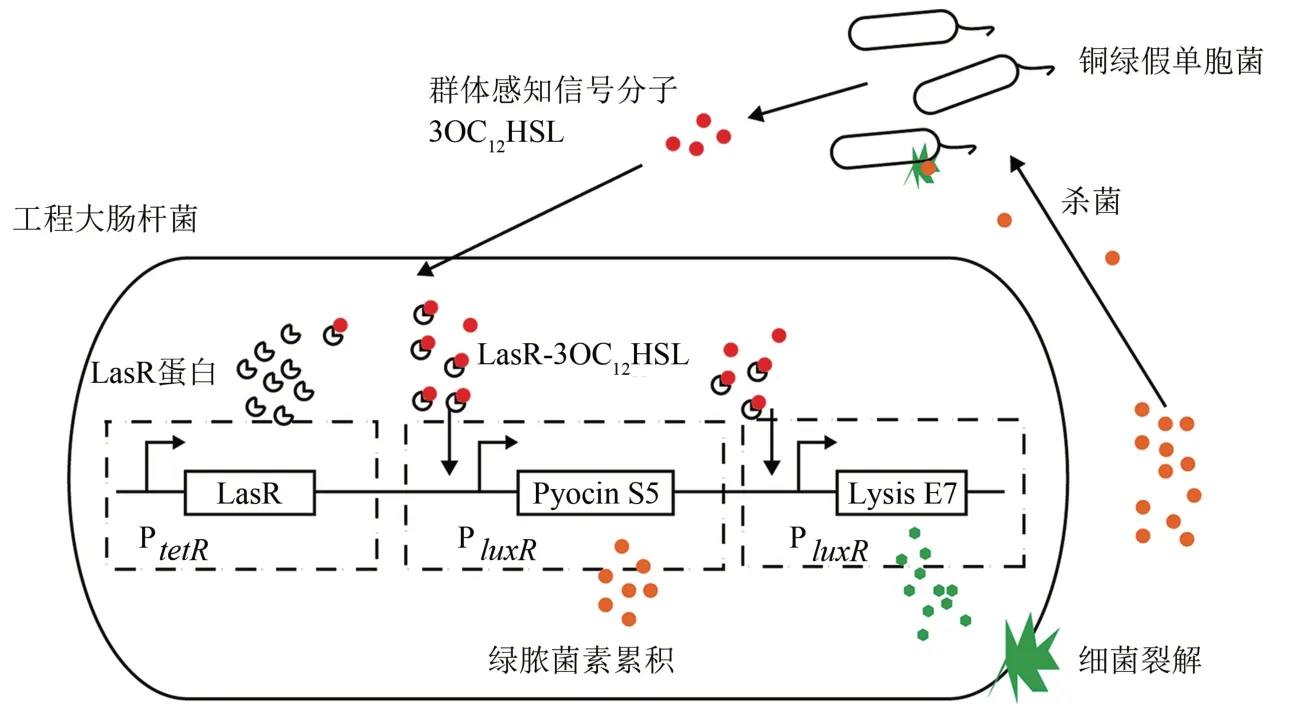
图6 感知并杀灭致病菌系统示意图[50]Fig.6 Schematic of the pathogen sensing and killing system[50]
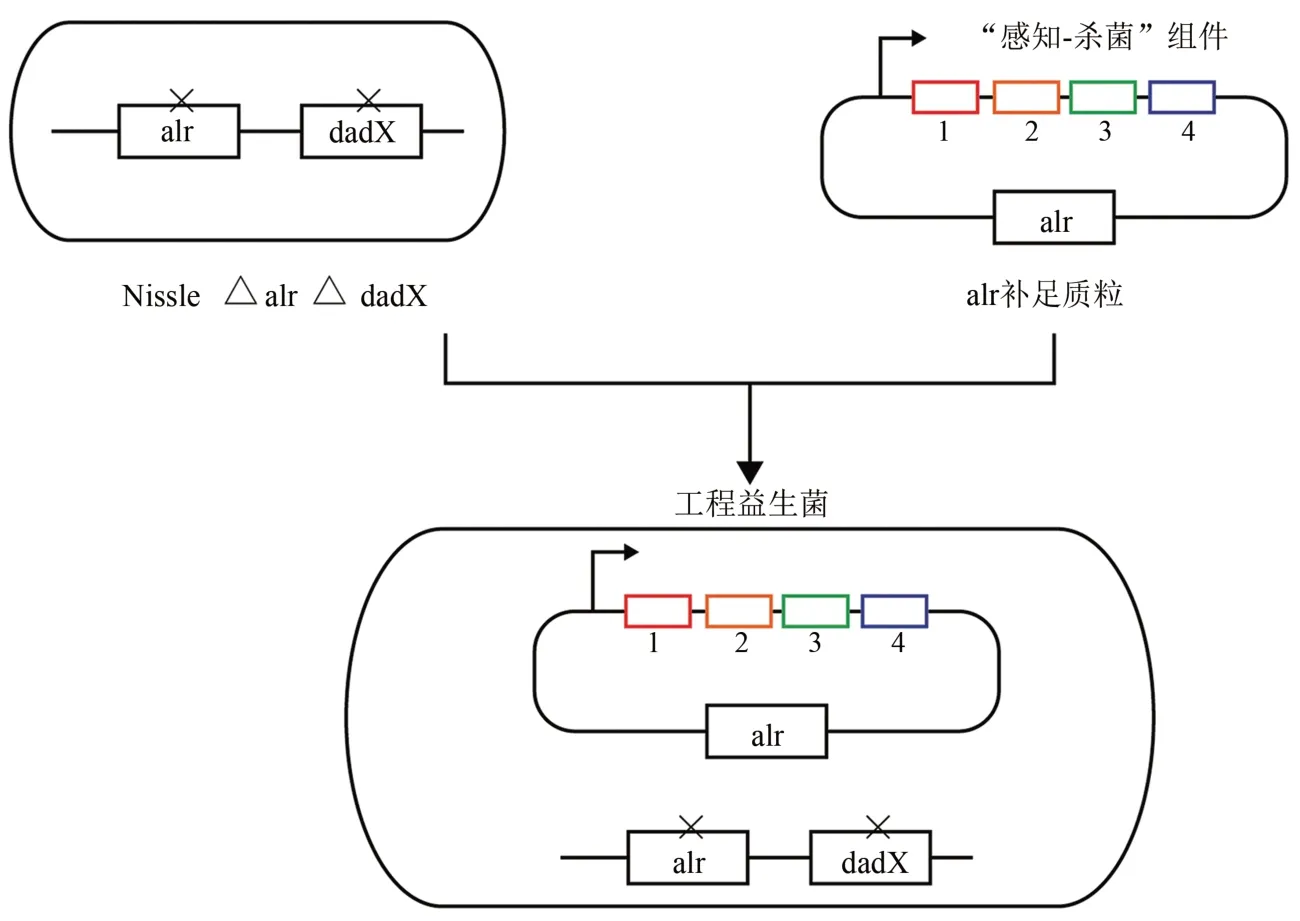
图7 治疗铜绿假单胞菌的工程益生菌大肠杆菌Nissle菌株示意图[56]Fig.7 Schematic of engineering the probiotic Nissle strain from E.coli for preventing and eliminating P.aeruginosa infection[56]
科学家们还发现CRISPR 系统参与到病原体细菌如弗朗西丝氏菌、铜绿假单胞菌、大肠杆菌等的致病过程中,辅助破坏宿主的免疫系统或涉及细菌耐药性的产生等。这些发现使我们对CRISPR系统作为细菌生理调节功能有了新的认识,进一步研究某些细菌病原体的CRISPR 系统的作用,可以让我们更详细地了解其毒性及致病机理,这可能为致病干预或治疗提供新的策略。
5 合成生物学在病毒感染性疾病治疗中的应用
合成生物学在病毒感染性疾病治疗中的应用,主要集中在对病毒感染性疾病致病机理的研究和基于CRISPR技术的治疗方案。
5.1 合成生物学在病毒感染性疾病致病机理研究中的应用
合成生物学的出现为研究病毒致病机理提供了一种新的视角。随着科学家们能够以更快的速度合成和装配病毒基因组[60]的序列,大大提高了我们对宿主-病原体相互作用和疾病机理的认知。合成生物学的原理之一“合成分析”[61],即通过将单个遗传组分的快速合成、装配、重组和突变与直接的功能分析结合起来,为病毒病理学致病机制的理解提供了一种新的思路。在研究病毒感染性疾病时,通过重构致病病毒基因组,来研究其致病机理。例如,利用从冷冻保存的组织样品中提取的基因片段的序列信息合成H1N1病毒的基因组,H1N1病毒曾造成全球5000万以上的人员死亡。科学家们通过重构病毒对基因进行功能性分析,挖掘出了两种H1N1 病毒致病性的主要毒力因子:一种可以在不激活胰蛋白酶情况下诱导膜融合的血凝素变体,另一种可以增强病毒复制能力的改良聚合酶[60]。研究还揭示了1918年大流感病毒的超强毒性与8种基因的联合作用有关[60-62]。这些研究可以帮助人们确定未来病毒变种的毒力分析和潜在威胁[63-64]。
针对嵌合病毒的合成和分析对我们认识造成2002—2003 年严重急性呼吸综合症(severe acute respiratory syndrome,SARS)的冠状病毒[65]作出了重大贡献。因为SARS冠状病毒的直系祖先不能在实验室模式动物中传播,所以对SARS冠状病毒发展进程的表征,尤其是对其宿主趋向性改变的表征,变得极具挑战性。为此,科学家们设计并合成了一个30 kb 包含了与人类同源的受体结合刺突蛋白的类SARS 蝙蝠冠状病毒[66],这一合成的嵌合病毒能够在培养基中复制并感染小鼠。刺突蛋白是SARS冠状病毒表面最重要的膜蛋白,承担了病毒与宿主细胞受体结合并担负膜融合功能,是各种冠状病毒诱导免疫反应的主要抗原。通过动物实验,确认了刺突蛋白是造成人畜共患冠状病毒宿主趋向性改变的主要因子之一,并且揭露了具有更强感染力的刺突蛋白突变体病毒株。
另外,DNA 合成重建病原体还可用于生产诊断性高密度抗原组[67-68],如用于分析后莱姆病综合征或对丙型肝炎病毒(hepatitis C virus,HCV)和艾滋病病毒(human immunodeficiency virus,HIV)[69]有体液免疫反应的抗原组。
5.2 CRISPR技术在病毒感染性疾病治疗中的应用
病毒由一个核酸分子与蛋白质构成,利用宿主的细胞系统进行自我复制,无法独立生长和复制。一些致病性病毒,如HIV、乙型肝炎病毒(hepatitis B virus,HBV)可以在人体内长时间存在,并且依然保持感染性且不受宿主免疫系统的影响,造成病毒持续感染(viral persistence)。近年来,CRISPR 技术在体外或动物模型中被用于减少或消除持续性病毒感染,为潜伏性和慢性病毒感染的治疗带来了新的希望。
HIV是造成人类免疫系统缺陷的一种病毒,通过直接侵犯人体的免疫系统,破坏细胞免疫和体液免疫,不仅使人体难以抵御病原体侵害,而且给特效治疗药物和疫苗的研制带来了困难。目前,感染了HIV 的病人主要依靠各种抗病毒药物进行治疗。但是,这种疗法并不能完全治愈病人,需要终身服药。HIV-1 是艾滋病病毒最常见和最致病的毒株。消灭HIV-1需要清除受感染细胞和组织中发生整合反应的前病毒DNA。研究人员开发了一种联合疗法,靶向一群受感染小鼠体内的HIV[70]。该疗法使用一种持续的药物递送系统和基于CRISPR-Cas9的基因编辑技术,前者会持续多天缓慢释放药物并抑制病毒活性,后者则会通过切断相关的DNA 片段,消除被感染细胞中的病毒遗传密码。经过连续的治疗,研究团队使用解巢式PCR(nested PCR)、微滴式数字PCR(digitaldroplet PCR)及RNAscope 技术,发现在接受治疗后的5 周内,已经无法在小鼠的血液、淋巴组织、骨髓及大脑中检测到HIV。这一试验结果有很大的发展前景,研究人员计划将在此基础上开展进一步的研究,改进针对病毒的药物递送,特异性地消除潜伏的病毒感染。
通常情况下,HIV 病毒主要通过CCR5基因编码白细胞上的一种受体进入细胞。如果个体携带两个CCR5突变拷贝,那么HIV 病毒株无法进入细胞。但这种抗HIV 基因特别罕见:仅1%的欧洲血统人口携带。我国北京大学邓宏魁教授、307 医院陈虎教授以及北京佑安医院吴昊教授研究组[71]合作,报道了首例利用CRISPR-Cas9 在造血干细胞和祖细胞(hematopoietic stem and progenitor cells,HSPCs)中编辑CCR5基因并成功移植到罹患HIV和急性淋巴细胞白血病的患者案例,移植治疗使病人的急性淋巴白血病得到完全缓解,携带CCR5突变的供体细胞能够在受体体内长期存活达19 个月,且未引起明显的副作用,但细胞数量较少,对抵制HIV 感染效果欠佳,需进一步研究。这项临床研究初步探索了CRISPR的可行性和安全性。
截至2019 年,病毒性肝炎仍是全球主要的公共卫生问题之一,其中乙肝和丙肝影响到全球3.25亿人,每年导致约140万人死亡。据统计,我国乙肝病毒感染者约8600 万人,丙肝感染者约1000 万人,每年约33 万人死于乙肝或丙肝感染导致的肝硬化和原发性肝癌。尽管针对HBV 研制的有效预防性疫苗已经投入使用几十年,但HBV 仍然是当今全球十大死因之一,这是由于目前的治疗药物无法清除或使HBV 在肝细胞核内稳定存在的闭合共价环状DNA(cccDNA)完全失活,在停止治疗或出现耐药性突变毒株后,HBV 可以继续以cccDNA 为模板,产生子代病毒,重新活化。由此,研究人员利用CRISPR/Cas9 系统,对受到感染的哺乳动物肝细胞进行基因组编辑。Chen 等[72]设计了8 种针对HBV 基因型A 的gRNA,在转染HBV 质粒的Huh7 细胞模型种鉴定出了2 个有效的gRNA,并在水流动力学注射HBV 质粒的小鼠模型种进一步证明了CRISPR/Cas9 系统可以切割肝内含有HBV 基因组的质粒,降低小鼠血清中HBV表面抗原水平。另一方面,CRISPR/Cas9系统也被用于对HCV的抑制中。Weiss和Grakoui等[73]通过重新编辑弗朗西丝菌Cas9 蛋白,可以靶向HCV 病毒,抑制HCV在人类细胞中的复制。
6 合成生物学在感染性疾病预防中的应用
合成生物学不仅参与感染性疾病的治疗、感染性疾病致病机理和传播机制的研究,同时在预防感染性疾病方面也取得了一些进展[64,74-76]。
6.1 工程细菌在感染性疾病预防中的应用
细菌之间可以通过化学物质联系,像我们熟知的群体感知[77]。单细菌产生和分泌信号分子,这些信号分子是多种细菌共有的,或者是种特异性的。这些分子随着细菌种群的增长而积累,并能与受体结合,从而协调整个菌落范围内的基因表达或操纵其他细菌种群的行为。由此,我们除了利用工程菌结合细菌间的竞争生存机制治疗感染性疾病,也可以利用其来预防感染性疾病。霍乱弧菌产生的霍乱自动诱导因子1(CAI-1)和自动诱导因子2(AI-2),是关键毒力因子的抑制开关。March 等[78]构建了一种工程益生大肠杆菌,这些菌株本身可以生产AI-2,在改造后可以持续合成CAI-1;给被霍乱弧菌感染的小鼠喂食这种工程益生菌可以显著提升小鼠的存活率。这种方法可能成为预防感染性疾病的一种经济型的策略。与抗生素不同,基于群体感知的干预并不杀死病原体,而是重塑它们的行为,这种策略因为没有选择压力的存在,不易使病原体产生抗性。
6.2 合成生物学在疫苗研发中的应用
免疫接种对于控制感染性疾病,保护人类和动物健康是一个非常有效的手段。合成生物学的出现对疫苗的研发有了新的突破,出现了多种不同的研究策略,为更加理性的设计更多安全性高且易于接种的疫苗提供了新的思路。
6.2.1 人工病毒在减毒疫苗构建中的应用
利用合成生物学原理,从遗传组分中进行全基因组的高通量、高精度组装和合成,为设计用作疫苗的减毒病原体提供了新的方法。例如,对高剂量选择性表达特定基孔肯雅病毒(CHIKV)结构蛋白而产生的类病毒颗粒产生免疫的灵长类动物,可以抵抗病毒血症;即使是用猴源抗体治疗的免疫缺陷小鼠也能在随后的致死剂量的CHIKV感染中存活下来[79]。
此外,DNA 合成和组装也在开发针对脊髓灰质炎病毒[80]的安全活疫苗方面发挥了重要作用。脊髓灰质炎病毒可以通过病毒衣壳基因中相邻密码子对从高到低表达的系统基因逐级变化而减弱,例如,与GCA|GAG 相比,GCC|GAA 的表达量明显不足,尽管两者都编码Ala-Glu。这些变化降低了病毒的转译量,削弱了病毒的复制能力和传染性。这种毒性减弱的脊髓灰质炎病毒给小鼠提供了保护性免疫,并且具有一个较高的安全标准,因为所有631个基因组个体变化发生逆转并重构成感染性野生型病毒的可能性很低。这种基因组工程方法可以代表针对感染性疾病设计活疫苗的一般策略。其他有潜力的疫苗接种例子如使用抗原产生免疫刺激脂质体[81]作为基因可编程的合成疫苗和生产热稳定的口服型的基于藻类的疫苗,用于抵抗金黄色葡萄球菌感染[82]。
北京大学周德敏教授课题组[83]通过合成生物学方法构建减毒活疫苗,开发了被称为“合成减毒病毒工程技术”的新策略。他们利用终止密码子可以识别非天然氨基酸的原理,将病毒复制基因的部分编码密码子突变成终止密码子,使其在感染人体细胞后,不能进行完整的蛋白质翻译,从而获得了甲型流感病毒的活病毒疫苗。所获得的病毒疫苗具有安全性和有效性。进一步突变3个以上三联密码子,使病毒由预防性疫苗变为治疗病毒感染的药物,且其药效随着三联密码子数目的增加而增强。与依赖于少数的几个氨基酸突变所获得的传统减毒活病毒相比较,合成减毒病毒的减毒效果来源于多达几百处的密码子变化所产生的累积效应,回复突变的风险极低,大大提高了疫苗的安全性。
我国中国科学院武汉病毒研究所郑振华研究员团队[84]成功利用合成减毒病毒工程技术研制出了新型的ZIKV 减毒疫苗,小鼠在单次免疫刺激后就可以产生高滴度中和抗体,获得完全的攻毒保护,并且可以阻止ZIKV 通过母体垂直传播给子代。弱毒疫苗的基因组中引入了2568个同义突变,因此几乎不可能回复突变到野生型病毒。
6.2.2 人工细菌在减毒疫苗构建中的应用
人工减毒细菌不仅可以作为疫苗,还被证实是理想的疫苗载体。以口服方式进行免疫接种,细菌在体内可以有限增殖,一次免疫即可提供保护。细菌载体在增殖过程中能够表达多种免疫原性物质,诱导产生黏膜免疫应答,大约90%的人类和动物传染病病原体感染是起始于黏膜表面的。此外,人工减毒细菌还能表达单一的或多重抗原,从而对一种或几种病原体的感染提供保护。现已知沙门氏菌[85]、李斯特菌[86]等细菌常选被作为疫苗或疫苗载体的基础菌种。
减毒的沙门氏菌活株具有作为疫苗载体的巨大潜力,沙门氏菌基于Ⅲ型分泌系统(T3SS)的跨膜通道可以用来释放跟其Ⅲ型毒性因子融合的外源蛋白。Hensel 等[87]研究了沙门氏菌致病性毒力岛编码的T3SS 的各种效应蛋白对模型抗原的传递和免疫应答的诱导作用。这些效应蛋白SifA、SteC、SseL、SseJ 和SseF 跨膜后有同一个内体膜相关的亚细胞定位信息,所有效应蛋白都可以与模型抗原卵清蛋白和李斯特菌溶蛋白一起转移到宿主细胞的胞浆中。在体外试验中,SseJ 和SteC融合蛋白对T 细胞的刺激反应表现优于SseF 融合蛋白;而在接种了沙门氏菌载体菌株的小鼠中,只有基于SseJ或SifA的融合蛋白才能引发有效的T细胞反应。由此,T3SS 介导的抗原传递,选择最优的效应蛋白应是合理设计有效的沙门氏菌疫苗的关键。
6.2.3 酵母改造在新型疫苗构建中的应用
酵母如酿酒酵母、毕赤酵母等,因其具有完整的基因组序列信息、稳定的遗传性、易于培养和非致病性,已经成为人类公共卫生或农场畜牧业疫苗开发的理想模型系统。酿酒酵母和毕赤酵母经常用于表达具有治疗感染性疾病作用的外源蛋白。如重组酵母乙肝疫苗[88]是一种乙肝表面抗原亚单位疫苗,它是采用转基因的方法将乙肝病毒表达表面抗原的基因进行质粒构建,转入进入酿酒酵母中,通过培养这种重组酵母菌来表达乙肝表面抗原亚单位,具有原料易得、产量大、安全、高效等特点。重组芽殖酵母[89]可以在小鼠肠道中分泌蛋白质或肽,为开发口服疫苗开辟了道路。针对病毒感染性疾病,基于酵母制备的疫苗研究还涉及针对HPV[90]、HCV[91]、肠道病毒[92]、DENV[93]、甲型流感病毒H1N1[94]等病毒病原体。针对细菌感染性疾病,基于酵母制备的疫苗研究涉及炭疽杆菌[95]、结核分岐杆菌[96]、李斯特菌[97]等细菌病原体。
6.3 合成生物学通过改造感染源限制疾病的传播
感染性疾病具有感染源,他们是病原体自然生存、繁殖并排出的宿主或场所。感染源的种类包括患者、病原体携带者、动物及某些场所的物品。昆虫是动物传染源中常见的感染源之一,科学家们利用带有条件显性致死合成回路的转基因病毒株抑制昆虫病媒种群,可能控制疟疾寄生虫和登革病毒的传播,并可能最终控制无法治疗的疾病的传播。Fu 等[98-100]改造了这种转基因的蚊子,蚊子体内含依赖四环素的反激活因子(tTA),这种基因只在雌蚊子的间接飞行肌中表达,并且只有在抑制这种基因转录,即四环素存在的情况下才能繁殖。而四环素的缺失造成一种特异性的雌性不能飞行表型的形成。将这种转基因蚊子的卵投放到生态系统中,只有雄性蚊子会释放到环境中;雌性蚊子仍然滞留在地面,不能进食、交配或吸血,这实际上是一种致命的表型。雄性蚊子不会传播这种疾病,但它们会将这种合成回路传播到原住民,即野生型蚊子种群中。
利用第一代tTA 转基因蚊子进行释放携带显性致死基因昆虫技术的野外测试早已在大开曼岛开展起来。首先,小规模投放一群转基因雄蚊证实其可以存活,与野生雌蚊交配并且产生转基因幼虫,接着大范围的试验结果显示,投放转基因雄蚊约11周后,野生蚊子的数量减少了80%。然而由于研究地点不是隔离的,且周围地区有高密度的野生型蚊子,因此实际的抑制效率仍然极具挑战性[75,101]。
类似的研究比如利用一个合成的基于归巢核酸内切酶的基因驱动系统传播改造的基因,将疟疾抗性基因从工程改造的蚊子传递给野外蚊子种群[102]。归巢内切酶通常在宿主基因组中产生一个单序列特异性双链断裂,然后以归巢内切酶基因(HEG)为模板进行同源重组修复。如此,自私的基因HEG在被称为“归巢”的基因转换过程中被复制到断裂的染色体上。在雄性种系启动子的控制下表达HEG I-SceI,可以使纯合子雄蚊高效归巢并快速产生遗传驱动,从而导致了被捕获的蚊种群的HEG 入侵。通过工程化其他HEG 的序列特异性,例如,I-AniI或I-CreI,利用基因驱动的概念,原则上可以用于敲入或敲除针对蚊子作为感染性疾病传染源能力的基因功能。
我国中国科学院上海植物生理生态研究所联合约翰霍普金斯大学彭博公共卫生学院[103],发现了一种能在按蚊中进行持续跨代传播的沙雷氏菌属(Serratia)新菌株AS1,并成功地人工构建出能同时分泌表达5个抗疟疾基因的菌株,能够有效减少92%~93%的疟疾原虫卵囊。通过高效驱动抗疟疾效应分子快速散播到整个蚊群中,能够使按蚊成为无效的疟疾媒介,实现从源头上阻断疟疾传播。
7 总结与展望
在人类与感染性疾病抗争的漫长岁月里,随着抗病原体药物的广泛大量使用,病原体耐药性的出现和耐药率的不断增高正成为全球人类热点关注的问题。病原体的耐药性不仅降低抗病原体药物的有效率,也严重威胁人类健康。合成生物学的出现为研究感染性疾病的机理、治疗及预防提供了新的思路,并已取得了阶段性的成果。
合成生物学不仅在感染性疾病治疗方面颇有应用,在治疗癌症、代谢系统疾病、再生医学等方面也有相当的应用。然而,要将以合成生物学为基础的生物医学疗法,真正应用到临床治疗中,我们仍然还有很长的路要走。合成生物学疗法临床化最重要的挑战之一是如果将合成的治疗回路、装置或有机体植入患者的特定细胞,并确保不会干扰人体的新陈代谢。同时,以合成生物学为基础的治疗方案的临床应用,与所有以基因和细胞为基础的治疗,都将面临相同的科学、伦理和法律问题,但由于合成生物学疗法提供更复杂的控制动力学,有望产生更高的治疗效果。虽然目前合成生物学疗法还处在试验阶段,还没有由合成生物学家开创的合成生物设备、人工基因网络或相关的产品用于临床治疗,但是合成生物学凭借自身优势,必将在21世纪生物医学发展中扮演重要角色。
猜你喜欢
传染病信息(2022年6期)2023-01-12 08:59:04
昆明医科大学学报(2022年2期)2022-03-29 00:51:20
中国动物传染病学报(2021年3期)2021-07-21 03:19:28
科学(2020年3期)2020-11-26 08:18:22
科学(2020年3期)2020-11-26 08:18:22
家庭医学(下半月)(2020年2期)2020-05-11 02:07:24
家庭医学(下半月)(2019年10期)2019-11-16 08:59:46
家庭医学(下半月)(2019年9期)2019-10-12 08:04:24
现代园艺(2017年13期)2018-01-19 02:27:58
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:44
