
利用酵母双杂交技术筛选鉴定与番茄环纹斑点病毒NSs蛋白互作的西花蓟马蛋白
2020-08-06 06:33赵星月陈建斌王曙光张晓林赵立华陈永对郑立敏
昆虫学报 2020年6期
赵星月, 陈建斌, 王曙光, 张晓林, 穆 野, 魏 辉, 赵立华, 陈永对, 郑 雪, 陈 勇, 郑立敏,*, 张 洁,*
(1. 西南林业大学生命科学学院, 昆明 650224; 2. 云南省农业科学院生物技术与种质资源研究所, 云南省农业生物技术重点实验室, 农业部西南作物基因资源与种质创制重点实验室, 昆明 650205; 3. 湖南省农业科学院植物保护研究所, 长沙 410125; 4. 福建省农业科学院植物保护研究所, 福建省作物有害生物监测与治理重点实验室, 农业部福州作物有害生物科学观测试验站, 福州 350003)
番茄环纹斑点病毒(tomato zonate spot virus, TZSV)是在云南省发现的布尼亚病毒科(Bunyaviridae)番茄斑萎病毒属Orthotospovirus暂定种,该病毒侵染危害烟草Nicotianatabacum、番茄Lycopersiconesculentum、辣椒Capsicumannuum和花卉等重要经济作物,近年来有向云南周边地区扩散蔓延的趋势(Dongetal., 2008)。TZSV侵染寄主植株后,呈现出典型的番茄斑萎病毒属细胞病理特征,病毒粒子分布于感染植物细胞的细胞质中,呈球形,由一层糖蛋白膜包裹,病毒粒子相对较大,直径约85 nm,单个或多个病毒粒子聚集于囊泡内。植物感染该病毒后,新叶首先出现黄色同心环纹或环形带状褪绿斑点,之后由黄斑转变为枯斑,叶片出现黄化、凋零,造成整叶或半叶呈红褐色坏死。感病植株还会出现矮化、顶芽萎蔫下垂、叶片皱缩褪绿等症状(Dongetal., 2008; 赵绕芬和王小兵, 2012)。虽然近年来许多学者对TZSV的病原生物学进行了广泛的研究,明确了病毒的地理分布、症状、病原性质、细胞病理学等诸多问题,但对TZSV编码蛋白与寄主植物和介体昆虫蛋白互作机制仍然知之甚少,尤其对该病毒在植物体内的复制增殖、运动、致病等作用机制以及与寄主互作相关研究欠缺。
TZSV病毒是一种RNA病毒,其基因组由3条RNA链组成,即ssRNA-L, ssRNA-M和ssRNA-S(Elliot, 1990; Germanetal., 1992)。其中,ssRNA-L是反向负义链,编码依赖RNA的RNA聚合酶(RdRP)(de Haanetal., 1991);早在20世纪70年代,RdRP就在病毒中被发现,大多数的RNA病毒都编码RdRP,主要参与病毒基因组的复制、mRNA合成、RNA重组等过程。RdRP在病毒复制中起着至关重要的作用。ssRNA-M编码非结构蛋白NSm(Kormelinketal., 1993);由RNA-M正义链编码的非结构蛋白NSm是唯一一种布尼亚病毒科其他动物病毒所不能编码的蛋白,被认为是Orthotospovirus适应植物的结果(Lietal., 2009)。Kormelink等(1994)首次提取和纯化了NSm,利用免疫标记等方法证明了NSm参与了病毒的胞间运动(cell-to-cell movement)。ssRNA-S编码非结构蛋白NSs(de Haanetal., 1990)。TZSV主要由蓟马以持久增殖型方式传播。本课题组前期调查发现,在TZSV感染作物发病区,西花蓟马Franiklinellaocicdentalis为优势种群,此外还发现有棕榈蓟马Franiklinellapalmi和梳缺花蓟马Franiklinellashculetzi(尹跃艳等, 2008; 郑雪等, 2015)。
NSs是ssRNA正链编码的非结构蛋白,最初被认为是病毒的致病因子,该蛋白在植物体内表达能够增强病症表型(Kormelinketal., 1991)。Takeda等(2002)首次用农杆菌共浸润实验证实了番茄斑萎病毒(tomato spotted wilt virus, TSWV)NSs蛋白为基因沉默抑制子。近年来的研究显示NSs可能在病毒复制、转录过程中起重要作用(Oliveiraetal., 2011; Margariaetal., 2014)。而关于TZSV NSs在病毒与介体昆虫互作中发挥的具体功能和机制尚不清楚。本研究利用酵母双杂交系统,以TZSV非结构蛋白NSs为诱饵蛋白,筛选西花蓟马体内与NSs互作的介体蛋白。通过对互作介体因子进行生物信息学分析,推测其在病毒与介体昆虫互作过程中可能具有的生物学功能,为深入了解TZSV致病特征及病原与介体昆虫的互作机制提供依据。
1 材料与方法
1.1 材料与试剂
本实验室感染TZSV和TSWV毒株采自云南元谋,经分离纯化获得的病毒汁液摩擦接种到本氏烟Nicotianabenthamiana,感病植株叶片保存于超低温冰箱作为毒源。西花蓟马的cDNA文库由湖南省农业科学院植物保护研究所馈赠。大肠杆菌EscherichiacoliDH5α为本实验室保存。克隆载体pEASY-T1、TransStart®TopTaqDNA聚合酶、High Pure dNTPs、10×Taq Buffer和琼脂糖等试剂购自TransGen Biotech有限公司;质粒小提试剂盒、酵母质粒提取试剂盒、溶壁酶购自天根生化科技(北京)有限公司;TRIzol购自Invitrogen公司;反转录试剂HiScript 1st Strand cDNA Synthesis Kit购自Vazyme Biotech有限公司;DNA限制性内切酶、营养缺陷型培养基、无氨基氮源、YeastmakerTMCarrier DNA Denatured、T4 DNA连接酶、酵母菌株AH109、pGADT7和pGBKT7质粒等试剂购自宝生物工程(大连)有限公司;胰蛋白胨和酵母提取物购自Oxoid公司;葡萄糖、二甲基亚砜(DMSO)、聚乙二醇(PEG)、醋酸锂(LiAc)、X-α-Gal等试剂购自Sigma公司;琼脂粉购自Difco公司;其余试剂为国产分析纯(A.R.)。
1.2 酵母双杂交诱饵质粒的构建
将1.1节约0.1 g感染了TZSV的本氏烟样品在液氮中充分研磨成粉末,迅速转移至液氮冷冻过的1.5 mL离心管中,加入1 mL Trizol,剧烈震荡混匀,然后参考Invitrogen公司Trizol试剂的操作方法提取总RNA;根据GenBank已登录的TZSVNSs(GenBank登录号: KC133530.1)序列设计用于PCR扩增TZSVNSs的特异引物,并根据酵母表达载体构建的要求,在上游引物和下游引物分别加入特定的酶切位点NdeⅠ和BamHⅠ(表1),引物序列见表1。以提取的总RNA为模板,NSs基因对应的3′端下游引物NSs-R进行反转录合成cDNA,反应体系: RNA 2 μg, NSs-R引物(2 μmol/L)1 μL, ddH2O至8 μL。反应条件: 65℃加热5 min,放置于冰上静置2 min,加入2×RT Mix 10 μL, HiScript® Enzyme Mix 2 μL, 50℃反应1 h,85℃灭活5 min,冰上放置即为获得的cDNA模板。最后以cDNA为模板,利用NSs基因的引物(NSs-F/NSs-R)进行PCR扩增获得目的基因,反应体系(50 μL): 10×buffer 5.0 μL,TopTaqDNA聚合酶 1.0 μL, dNTP Mix 4.0 μL, NSs-F和NSs-R(2 μmol/L)各1.0 μL, cDNA模板5.0 μL, ddH2O 33.0 μL。反应程序: 95℃预变性5 min; 95℃变性30 s, 58℃退火30 s, 72℃延伸1.5 min, 共30个循环; 72℃延伸10 min。PCR产物经1%琼脂糖凝胶电泳检测,进行胶回收并纯化。将PCR产物割胶回收后与克隆载体pEASY-T1连接,转化至大肠杆菌DH5α感受态细胞,涂布于含有50 μg/mL氨苄青霉素LB平板,菌落PCR及序列测定获得阳性克隆,命名为pEASY-NSs。用NdeⅠ和BamHⅠ分别消化重组质粒pEASY-NSs及pGBKT7载体质粒,纯化回收后,T4 DNA连接酶连接,转化至大肠杆菌DH5α感受态细胞,卡那霉素抗性平板筛选诱饵载体阳性克隆,菌落PCR和酶切鉴定获得诱饵载体的阳性克隆,命名为pGBKT7-NSs。将上述获得的质粒送往擎科生物技术有限公司进行测序,并通过NCBI网站的BLAST对测序结果进行比对,将测序结果完全正确的重组质粒扩大培养并保存备用。
1.3 酵母双杂交诱饵载体的毒性和自激活检测
采用醋酸锂法将诱饵表达载体pGBKT7-NSs、空载体pGBKT7(阴性对照组)分别转化至酵母感受态细胞AH109,并涂布于SD/-Trp上,30℃下倒置培养2~3 d后,观察菌落生长情况。同样采用醋酸锂法将阳性对照(pGBKT7-53/pGADT7-T)、阴性对照(pGBKT7-Lam/pGADT7-T)以及pGBKT7-NSs/pGADT7-T分别转化至酵母细胞AH109内,并涂布于SD/-Trp/-Leu培养基上,30℃下倒置培养2~3 d后,选取单克隆划线于SD/-His/-Leu/-Trp/-Ade/X-α-Gal和SD/-His/-Leu/-Trp/-Ade和SD/-Trp/-Leu营养缺陷型培养基上,30℃下倒置培养2~3 d,观察并比较酵母菌落的生长状态和颜色变化,判断NSs蛋白对酵母AH109细胞是否有毒性和自激活活性。
1.4 酵母双杂交顺序转化筛选与NSs互作的蛋白和生物信息学分析
采用顺序转化法筛选西花蓟马cDNA文库中与NSs互作的蛋白。将pGBKT7-NSs转化至酵母AH109后,以含有pGBKT7-NSs的AH109酵母菌制备感受态细胞。将西花蓟马的cDNA文库质粒转化至含有pGBKT7-NSs的AH109酵母菌感受态细胞中,涂布于含有2.5 mmol/L 3氨基-1,2,4三氮唑(3-AT)的筛选培养基SD/-His/-Leu/-Trp上,30℃倒置培养3~5 d后,挑取SD/-His/-Leu/-Trp+3-AT平板上生长的单菌落,接种于SD/-Ade/-His/-Leu/-Trp/X-α-Gal平板培养3~5 d,观察酵母的生长及显色情况。
挑取SD/-Ade/-His/-Leu/-Trp/X-α-Gal平板上的阳性菌落,加入到含有氨苄青霉素的液体SD/-Trp/-Leu培养基中,置于30℃, 220 r/min摇床培养24 h,参考酵母质粒小量提取试剂盒说明书提取质粒。以质粒为模板,pGADT7载体通用引物见表1。进行PCR扩增反应,分析pGADT7载体中插入片段的大小并排除重复克隆。舍弃插入片段小于500 bp的质粒,其余转化至大肠杆菌DH5α感受态细胞,送交公司测序。根据序列信息分析插入片段序列,参照NCBI数据库进行BLAST分析,找到同源序列,统计序列长度、基因登录号、基因注释、读码框完整性、读码框一致性等信息,初步筛选出与NSs互作的介体蛋白。
1.5 酵母细胞内回转验证TZSV NSs与介体蛋白候选因子的相互作用
将类电压依赖性阴离子通道(voltage-dependent anion-selective channel-like, VDAC)全长基因序列构建至pGADT7载体进行互作验证。根据GenBank已登录的西花蓟马VDAC(GenBank登录号: XM_026416489.1)序列设计VDAC的特异引物,并根据酵母表达载体构建的要求,在上游引物和下游引物分别加入特定的酶切位点NdeⅠ和BamHⅠ,引物序列见表1。取约20头西花蓟马,提取其总RNA,并以VDAC-R为特异性引物进行反转录,获得西花蓟马cDNA模板(RNA提取和反转录过程参考1.2节)。以VDAC-F/VDAC-R为引物,PCR扩增VDAC基因,反应体系和反应程序同1.2节。将扩增并纯化的VDAC基因扩增序列克隆到pEASY-T1载体上,经NdeⅠ和BamHⅠ双酶切后连接到酵母表达载体pGADT7,获得酵母重组表达载体pGADT7-VDAC。
将pGBKT7-NSs和候选因子pGADT7-VDAC共转化至酵母AH109感受态细胞,涂布于SD/-Trp/-Leu平板,30℃下倒置培养,直至克隆菌落出现,牙签蘸取SD/-Trp/-Leu平板上生长的单菌落,置于20 μL ddH2O中,即为初始菌液浓度100,按4个浓度梯度(10-1, 10-2, 10-3和10-4)对初始菌液进行稀释,分别接种于SD/-Trp/-Leu/-His/-Ade平板,同时设置阳性对照组(pGBKT7-p53/pGADT7-T)、阴性对照组(pGBKT7-Lam/pGADT7-T)、空载体共转化对照组(pGADT7/pGBKT7)以及质粒与空载体共转化对照组(pGBKT7-NSs/pGADT7; pGADT7-VDAC/pGBKT7)。30℃倒置培养3~5 d后,观察酵母的生长情况。
1.6 pGEX-4T-1-VDAC和pDEST17-NSs原核表达载体的构建
根据GenBank已登录的西花蓟马VDAC(GenBank登录号: XM_026416489.1)序列设计VDAC基因引物,如表1所示。以1.5节中获得的西花蓟马cDNA为模板为模板,PCR扩增VDAC基因,反应体系和反应程序同1.2节。将扩增并纯化的VDAC基因扩增序列克隆到pEASY-T1载体上,经EcoRⅠ和XhoⅠ双酶切后连接到pGEX-4T-1(带有GST标签蛋白)表达载体,获得与GST融合的VDAC原核表达载体pGEX4T-1-VDAC。
利用Gateway技术构建重组质粒pDEST17-NSs,设计引物如表1所示。以GW-TZSV-NSs-F/GW-TZSV-NSs-R为引物,以1.2节合成的感染了TZSV的本氏烟样品cDNA为模板,PCR扩增TZSV NSs基因并纯化回收,PCR反应体系和反应条件同1.2节。根据TSWV NSs基因序列(GenBank登录号: JF960235.1),设计TSWV NSs引物,引物见表1。提取感染TSWV植株总RNA,以GW-TSWV-NSs-R为特异性引物,反转录获得cDNA(方法同1.2节),以GW-TSWV-NSs-F/GW-TSWV-NSs-R为引物,PCR扩增TSWV NSs基因并纯化回收, PCR反应体系和反应条件同1.2节。
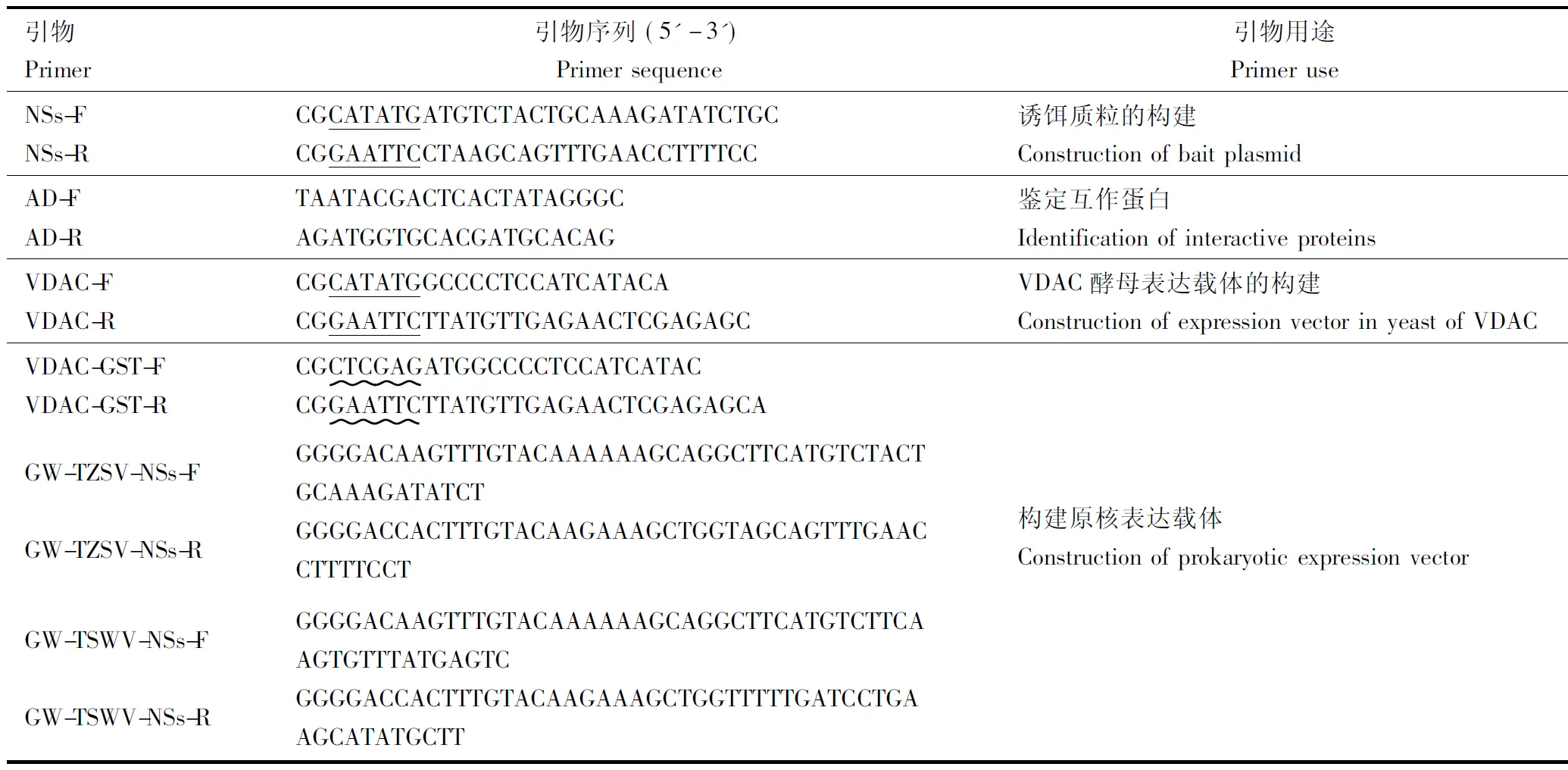
表1 引物信息Table 1 Primers used in this study
将上述TSWV NSs和TZSV NSs基因的PCR回收产物分别与pDONR221混合进行BP重组反应,反应体系: PCR产物1.5 μL, pDONR221质粒0.5 μL, BP重组酶0.5 μL,充分混匀后离心,置于25℃金属浴中反应1 h,转化大肠杆菌DH5α感受态细胞,分别获得来源于TZSV和TSWV的重组入门质粒载体pDONR221-NSs,并与原核表达载体pDEST17(带有His标签)进行LR重组反应,反应体系: pDONR221-NSs质粒0.5 μL, pDEST17质粒0.5 μL, BP重组酶0.5 μL, ddH2O 1 μL, 充分混匀后离心, 置于25℃金属浴中反应过夜,转化大肠杆菌DH5α感受态细胞,分别获得来源于TZSV和TSWV的重组质粒pDEST17-NSs。重组质粒经测序验证序列正确后,分别转化到大肠杆菌BL21(DE3)感受态细胞,氨苄霉素培养基筛选,获得pDEST17-NSs阳性菌落。
1.7 西花蓟马VDAC, TZSV NSs和TSWV NSs融合蛋白的诱导表达
pGEX-4T-1-VDAC和分别来源于TZSV和TSWV的pDEST17-NSs质粒转化大肠杆菌BL21(DE3)感受态细胞,挑取单菌落,分别接种于100 mg/L Amp和50 mg/L Kan的LB液体培养基中培养,0.5 mmol/L IPTG诱导表达融合蛋白。离心收集菌体,重悬于PBS,超声破碎后离心,上清、沉淀及对照样品(pGEX-4T-1空载体,仅表达GST蛋白)进行SDS-PAGE电泳分析。
1.8 GST Pull-down技术验证NSs蛋白与VDAC蛋白的相互作用
向装有谷胱甘肽琼脂糖4B(glutathione sepharose 4B)的纯化柱中加入蛋白结合缓冲液(50 mmol/L Tris-HCl, pH 7.4, 100 mmol/L NaCl),平衡5个柱体积。加入1.7节原核表达的VDAC-GST大肠杆菌裂解液,以表达GST蛋白的菌裂解液作为对照。放置于4℃水平振荡仪上充分振荡2~4 h后,用预冷的PBS缓冲液冲洗5~8次,弃掉非结合蛋白和杂蛋白。再将1.7节NSs-His大量诱导的菌裂解液(超声破碎,离心过滤后)分别加入结合GST-VDAC蛋白和GST蛋白的谷胱甘肽琼脂糖4B悬浮液中。同时设TSWV的NSs为对照蛋白,同样4℃水平振荡仪上缓慢振荡2~4 h。PBS冲洗5~8次,弃掉非结合蛋白,洗脱缓冲液(50 mmol/L Tris-HCl, pH 7.4, 10 mmol/L NaCl, 10 mmol/L还原性谷胱甘肽)洗脱纯化柱,收集样品,用于SDS-PAGE和Pull-down实验分析。用抗His抗体检测VDAC-GST和NSs-His的互作情况。抗GST抗体检测VDAC-GST和GST表达情况。
2 结果
2.1 酵母双杂交诱饵载体pGBKT7-NSs
以TZSV侵染植株总RNA反转录后的cDNA为模板,PCR扩增TZSVNSs。如图1所示,获得1 380 bp的特异性片段,与预期TZSVNSs片段大小一致。扩增产物构建至pEASY-T1克隆载体,获得pEASY-NSs,经NdeⅠ/BamHⅠ双酶切验证,得到3 928 bp的载体片段和1 380 bp的TZSVNSs片段,插入片段大小与目的基因大小一致(图2: A)。通过NdeⅠ/BamHⅠ双酶切pEASY-NSs和pGBKT7,将TZSVNSs构建至酵母表达载体pGBKT7上,双酶切鉴定,得到7 300 bp左右的载体片段和1 380 bp的TZSVNSs片段(图2: B),测序结果分析无误,表明成功构建了酵母表达载体pGBKT7-NSs。
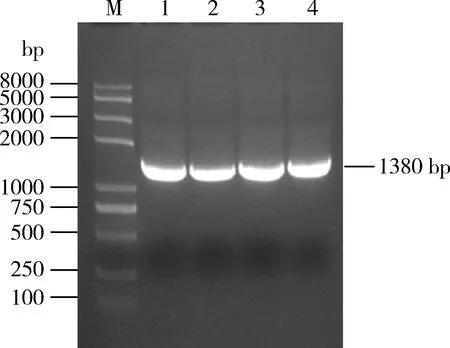
图1 PCR扩增TZSV NSsFig. 1 PCR amplification product of TZSV NSs M: Trans2K Plus Ⅱ DNA marker; 1-4: PCR产物PCR products.

图2 pEASY-NSs克隆质粒(A)和pGBKT7-NSs重组质粒(B)的双酶切鉴定Fig. 2 Double enzyme digestion of the cloning plasmid pEASY-NSs (A) and the recombinant plasmid pGBKT7-NSs (B) M: Trans2K Plus Ⅱ DNA marker; 1: pEASY-NSs克隆质粒的酶切产物Restriction enzyme digestion product of the cloning plasmid pEASY-NSs; 2: pEASY-T1克隆质粒的酶切产物Restriction enzyme digestion product of the cloning plasmid pEASY-T1; 3: pGBKT7质粒的酶切产物Restriction enzyme digestion product of the plasmid pGBKT7; 4, 5: pGBKT7-NSs重组质粒的酶切产物Restriction enzyme digestion product of the recombinant plasmid pGBKT7-NSs.
2.2 TZSV NSs蛋白对酵母AH109细胞毒性和自激活活性
比较转化有诱饵载体pGBKT7-NSs及空载体pGBKT7的酵母AH109细胞,发现两组菌落均为白色或棕色,且大小、数量均匀一致(图3),表明NSs蛋白表达对酵母AH109细胞无毒性。比较分别转化有pGBKT7-NSs/pGADT7-T,阳性载体pGBKT7-p53/pGADT7-T,以及阴性载体pGBKT7-Lam/pGADT7-T的酵母AH109细胞在SD/-His/-Leu/-Trp/-Ade/X-α-Gal、SD/-His/-Leu/-Trp/-Ade和SD/-Trp/-Leu培养基上的生长情况,发现转化有pGBKT7-NSs/pGADT7-T的酵母AH109细胞仅在SD/-Trp/-Leu培养基上生长(图4: C),而阳性质粒都能够在上述3种培养基上生长(图4: A-C),并在SD/-His/-Leu/-Trp/-Ade/X-α-Gal培养基上显现蓝色(图4: A),表明NSs蛋白在AH109细胞内无自激活活性。以上结果表明可用酵母双杂交筛选与TZSV NSs蛋白互作的西花蓟马蛋白。

图3 pGBKT7-NSs诱饵质粒对AH109酵母细胞的毒性Fig. 3 Toxicity of the bait plasmid pGBKT7-NSs to AH109 yeast cellsA: pGBKT7-NSs在SD/-Trp培养基上的生长情况Colony growth of pGBKT7-NSs on the medium SD/-Trp; B: pGBKT7在SD/-Trp培养基上的生长情况Colony growth of pGBKT7 on the medium SD/-Trp.
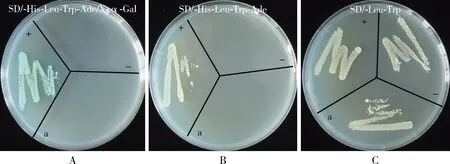
图4 pGBKT7-NSs诱饵质粒对AH109酵母细胞的自激活Fig. 4 Autoactivation of the bait plasmid pGBKT7-NSs to AH109 yeast cellsA: SD/-His-Leu-Trp-Ade/X-α-Gal; B: SD/-His-Leu-Trp-Ade; C: SD/-Leu-Trp. +: 阳性对照Positive control pGBKT7-53/pGADT7-T; -: 阴性对照Negative control pGBKT7-Lam/pGADT7-T;A: pGBKT7-NSs/pGADT7-T.
2.3 酵母双杂交筛选与TZSV NSs互作的西花蓟马蛋白
将西花蓟马cDNA文库质粒转化到含有诱饵质粒pGBKT7-NSs的AH109感受态细胞,观察菌落生长及变蓝情况(图5)。挑取菌落生长状况变蓝的菌落,接种至加有氨苄青霉素的SD/-Trp/-Leu液体培养基中扩繁并提取酵母质粒,PCR鉴定cDNA插入片段的大小。将扩增大于500 bp的酵母质粒转化至大肠杆菌细胞,测序并进行BLAST分析。 通过序列比对发现阳性克隆主要为VDAC(约1 000 bp)(GenBank登录号: XM_026416489.1)和缺失突变5′端序列突变体VDAC-M基因(约600 bp)(图6)。

图5 西花蓟马cDNA文库质粒转化含有诱饵质粒 pGBKT7-NSs的AH109酵母细胞的培养物Fig. 5 Culture of the transformation of cDNA library plasmid of Franiklinella ocicdentalis into AH109 yeast cells containing the bait plasmid pGBKT7-NSs
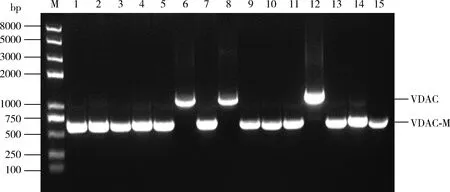
图6 酵母双杂交筛选西花蓟马cDNA文库中阳性克隆质粒的PCR鉴定Fig. 6 PCR identification of positive clones in cDNA library of Franiklinella ocicdentalis obtained by yeast two-hybrid screening VDAC: 类电压依赖性阴离子通道Voltage-dependent anion-selective channel-like. VDAC-M: 缺失突变5′端序列的VDAC突变体The 5′terminal deletion mutant of VDAC. M: Trans2K Plus Ⅱ DNA marker; 1-5, 7, 9-11, 13-15: VDAC-M; 6, 8, 12: VDAC.
2.4 酵母细胞内TZSV NSs与西花蓟马VDAC的相互作用
为了进一步验证TZSV NSs和西花蓟马蛋白VDAC的相互作用,本研究将pGBKT7-NSs和pGADT7-VDAC重新共转化酵母AH109感受态细胞,涂布平板,发现pGBKT7-NSs/pGADT7-VDAC实验组及阳性对照组的酵母生长情况较好(图7: A, C),其他对照组酵母没有生长迹象(图7: B, D-F)。这表明了西花蓟马VDAC蛋白和TZSV NSs在酵母中存在相互作用。
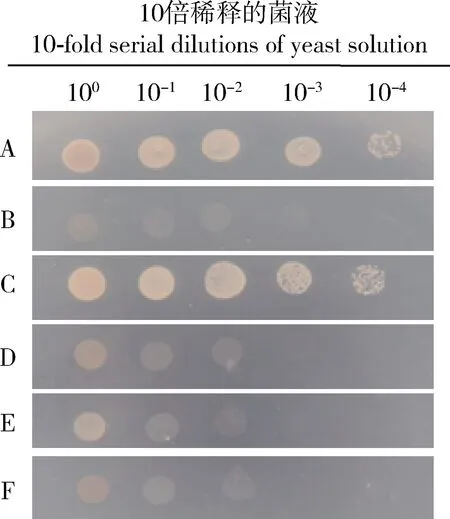
图7 pGBKT7-NSs和pGADT7-VDAC共转化AH109 酵母细胞的培养物Fig. 7 Culture of the cotransformation of pGBKT7-NSs and pGADT7-VDAC into AH109 yeast cellsA: 阳性对照Positive control (pGBKT7-p53/pGADT7-T); B: 阴性对照Negative control (pGBKT7-Lam/pGADT7-T); C: pGBKT7-NSs/pGADT7-VDAC; D: pGBKT7/pGADT7-VDAC; E: pGBKT7-NSs/pGADT7; F: pGBKT7/pGADT7. VDAC: 类电压依赖性阴离子通道Voltage-dependent anion-selective channel-like.
2.5 GST Pull-down体外验证NSs与西花蓟马VDAC的相互作用
GST Pull-down结果显示,在VDAC-GST和NSs-His共同沉淀组中检测到条带,与预计蛋白大小一致;而阴性对照组未检测到条带(图8),说明TZSV的NSs-His能够与VDAC-GST在体外结合,而不能与阴性对照GST结合,同时证明VDAC-GST不能够与阴性对照TSWV的NSs-His结合。 由此得出西花蓟马VDAC与TZSV的NSs存在体外特异性相互作用。
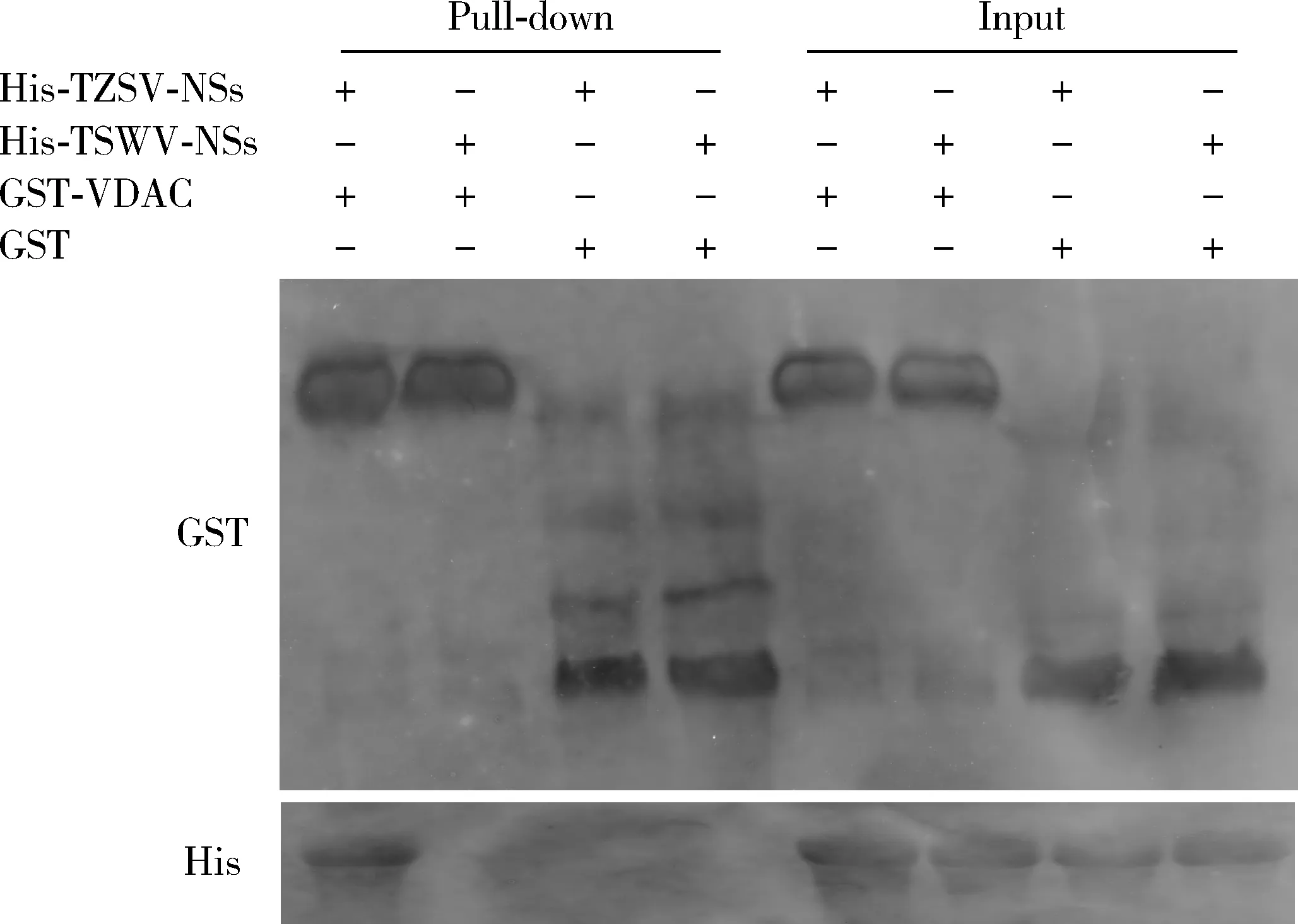
图8 GST Pull-down验证TZSV NSs与西花蓟马VDAC的体外互作关系Fig. 8 Interaction in vitro between TZSV NSs and VDAC of Franiklinella ocicdentalis verified by GST Pull-down Pull-down: Pull-down蛋白Pull-down protein; Input: 大肠杆菌总蛋白Total protein from cell extracts of Escherichia coli. +: 互作Interactive; -: 非互作Non-interactive.
3 讨论
本研究成功构建了NSs蛋白的酵母诱饵表达载体pGBKT7-NSs,用酵母双杂交系统从西花蓟马cDNA文库中初步筛选到与NSs互作的介体蛋白类电压依赖性阴离子通道(VDAC)。为了进一步确定NSs和VDAC之间相互作用,本研究通过GST Pull-down技术对TZSV NSs与VDAC的相互作用进行了体外验证。大肠杆菌内IPTG诱导VDAC-GST和对照GST蛋白,并纯化后捕获NSs-His蛋白,洗脱后经SDS-PAGE和Pull-down分析结果表明,TZSV NSs与VDAC蛋白在体外存在相互作用。VDAC是一种具有高度保守性的线粒体外膜孔状蛋白,也是一种具有电压依赖性和阴离子选择性的嵌入蛋白,通常具有β-折叠构成的跨膜结构,由10个氨基酸组成,广泛地存在于真核生物中(Schulz, 2000; Godboleetal., 2003)。VDAC位于线粒体的外膜,是线粒体代谢物进出的屏障,控制线粒体与其他部分的交互作用,在线粒体膜上形成亲水性通道,控制着线粒体内物质的进出,维持线粒体的稳态,VDAC的异常则会引起线粒体功能的紊乱(Bay and Court, 2002)。VDAC的表达水平升高或是降低均可使线粒体功能受到影响,在细胞生存中起着重要作用(Abu-Hamadetal., 2006; Shoshan-Barmatzetal., 2010)。例如,Vander-Heiden等(2000)在心衰动物模型和心衰患者的心脏中均发现了心肌细胞凋亡,同时检测到VDAC表达的下降。Sampson等(2001)研究发现,敲除了VDAC基因的雄性小鼠的精子细胞活力明显下降。VDAC下调导致细胞凋亡的机制尚不清楚,推测可能原因是VDAC的减少抑制线粒体功能,导致线粒体膜通透性发生改变,释放蛋白如细胞色素C、Smac/Diablo、细胞凋亡因子等最终导致凋亡(Green and Kroemer, 2004)。
除了调节线粒体的代谢和能量功能外,VDAC似乎是各种细胞存活和细胞死亡信号的汇聚点,这些信号由其与各种配体和蛋白质的关联介导(戴琼艳和段满林, 2015)。例如,脑神经药物多巴胺可使神经细胞线粒体VDAC的mRNA表达水平及蛋白表达水平降低,ATP水平也降低,诱导凋亡发生(Premkuar and Simantov, 2002)。因此推测,VDAC与TZSV NSs的互作关系成正相关关系,VDAC的下调可能会导致细胞的凋亡。故VDAC与TZSV NSs互作机制有待进一步研究,这不但有助于揭示TZSV NSs的分子作用机制,也能为研究VDAC的生物学功能奠定基础。
猜你喜欢
昆虫学报(2022年9期)2022-10-18
植物保护(2022年5期)2022-10-13
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
今日农业(2021年11期)2021-11-27
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
名人传记·财富人物(2017年9期)2017-11-02
名人传记·财富人物(2017年9期)2017-11-02
Coco薇(2016年8期)2016-10-09
