
思茅山橙中的单萜吲哚生物碱类成分(英文)
2020-08-03 00:38何隽张发雷望石琴王淙薇崔琴丹冯涛
中南民族大学学报(自然科学版) 2020年4期
何隽,张发雷,望石琴,王淙薇,崔琴丹,冯涛
(中南民族大学 药学院,武汉 430074)
Plants of the genusMelodinus(Apocynaceae) have a long history in folk use in China, and their chemical compositions and bioactivities are concerned by domestic and abroad scholars[1].M.henryiis a member of the family ofMelodinusplants,previous studies reported the isolation and cytotoxic activities of melohenines A and B,melodinines A-G, melodinine V, and melodinhenines A-F fromM.henryi[2-5].In order to elucidate the pharmacodynamic basis ofM.henryi, the chemical constituents ofM.henryihave been systematically studied, which led to the isolation of six alkaloids. The structures of them were identified. All compounds were obtained from this plant for the first time. In addition, all compounds were evaluated for their cytotoxicities against five human cancer cell lines.
1 Experimental section
1.1 Plant Material
The plants ofM.henryiwere collected from Mengla County, Yunnan province. It was identified by Mr. Jing-Yun Cui, Xishuangbanna Tropical Plant Garden. A voucher specimen(No. Cui20081128) has been deposited at South-Central University for Nationalities.
1.2 Experimental Instruments and Materials
UV spectra were obtained using a Double Beam Spectrophotometer UH5300(Hitachi High-Technologies, Tokyo, Japan).IR spectra were obtained by a Shimadzu IRTracer-100 spectrometer using KBr pellets.1D and2D NMR spectra were run on a Bruker Avance III 600 MHz spectrometer with TMS as an internal standard. Chemical shifts(δ) were expressed in ppm with reference to solvent signals. High resolution electrospray ionization mass spectra(HRESIMS) were recorded on a LC-MS system consisting of a Q ExactiveTMOrbitrap mass spectrometer with a HESI ion source(ThermoFisher, Germany) used in ultra-high resolution mode(140 000, atm/z200) and UPLC system(Dionex UltiMate 3000 RSLC, ThermoFisher, Germany).Column chromatography(CC)was performed on silica gel(200-300 mesh, Qingdao Marine Chemical Ltd.,), RP-18 gel(20-45 μm, Fuji Silysia Chemical Ltd., Japan), and Sephadex LH-20(Pharmacia Fine Chemical Co., Ltd., Sweden). Preparative High Performance Liquid Chromatography(prep-HPLC) was performed on an Agilent 1260 liquid chromatography system equipped with Zorbax SB-C18 columns(5 μm, 9.4 mm × 150 mm or 21.2 mm × 150 mm) and a DAD detector. Fractions were monitored by TLC(GF254, Qingdao Haiyang Chemical Co., Ltd), and spots were visualized by Dragendorff’s reagent.
1.3 Extraction and Isolation
The powdered plants ofM.henryi(40.0 kg) were extracted three times with 90% EtOH. The combined extracts were concentrated under reduced pressure, and adjusted to pH=2~3 with 5‰ HCl. The acidic mixture was defatted with ethyl acetate(EtOAc) and then basified to pH=9~10 with 10% ammonia solution. The aqueous phase was subsequently extracted with EtOAc to give an alkaloidal extract(201.3 g). The crude alkaloids was then subjected to a silicagel column(200-300 mesh) using CHCl3-MeOH gradient(1∶0-0∶1) to obtain five fractions(I-V). Fraction Ⅲ(2.1 g) was separated by silica gel CC(petroleum ether-Me2CO, 50∶1-5∶1) to yield three subfractions Ⅲ-1-Ⅲ-3.Subfraction Ⅲ-3 was purified on a preparative C18HPLC column with a gradient of ACN-H2O(48∶52) to yield1(3.2 mg). Fraction Ⅳ(5.2 g) was separated by silica gel CC(petroleum ether-Me2CO, 20∶1) to yield three subfractions Ⅳ-1-Ⅳ-3.Subfraction Ⅳ-1 was purified on a preparative C18HPLC column with a gradient of ACN-H2O(35∶65) to yield6(6.0 mg). Subfraction Ⅳ-2 was purified on a C18HPLC column with a gradient of ACN-H2O(28∶72) to yield2(3.4 mg). Fraction Ⅴ was separated by silica gel CC(CHCl3-MeOH, 20∶1-1∶1) to yield four subfractionsⅤ-1-Ⅴ-4.SubfractionⅤ-2 was purified on a C18HPLC column with a gradient of ACN-H2O(15∶85) to yield4(5.4 mg). Subfraction Ⅴ-3 was separated by silica gel CC(CHCl3-MeOH, 50∶1-5∶1) to yield four subfractionsⅤ-3-1-Ⅴ-3-4. Ⅴ-3-2 was purified on a C18HPLC column with a gradient of ACN-H2O(35∶65) to yield5(1.0 mg). Ⅴ-3-3 was purified on a C18HPLC column with a gradient of ACN-H2O(18∶82) to yield3(7.2 mg).
1.4 Cytotoxicity Assays
Five human cancer cell lines, human myeloid leukemia HL-60, hepatocellular carcinoma SMMC-7721, lung cancer A-549, breast cancer MCF-7, and colon cancer SW480 cells, were used in the cytotoxic assay. All the cells were cultured in RPMI-1640 or DMEM medium(Hyclone, USA), supplemented with 10% fetal bovine serum(Hyclone, USA) in 5% CO2at 37 °C. The assays were performed according to the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] method in 96-well microplates. Briefly, 100 μL of adherent cells was seeded into each well of 96-well cell culture plates and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition with aninitial density of 1 × 105cells/mL. Each tumor cell line was exposed to the test compound at concentrations of 0.064, 0.32, 1.6, 8, and 40 μM in triplicates for 48 h, with cisplatin(Sigma, USA) as a positive control. After each compound treatment, cell viability was detected and a cell growth curve was graphed. IC50values were calculated by Reed and Muench’s method.
2 Results and discussion
All alkaloids were identified by comparison of their NMR spectroscopic data with the literature,namely 19R-acetoxytabersonine(1)[6],11-methoxy-19-hydroxytabersonine(2)[7], 16β-hydroxy-19R-vindolinine(3)[8],19R-kitramine(4)[9], trichophylline(5)[10], voaharine(6)[11,12]. The structures of these compounds are shown in Fig.1. The structure with the absolute configuration of6was further determined by the single crystal X-ray diffraction, as shown in Fig. 2 [Flack parameter=0.06(5)].
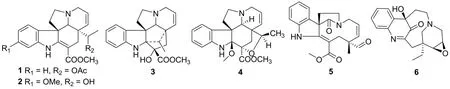
Fig.1 Structures of compounds 1 to 6

Fig.2 ORTEP drawing for 6 showing the absolute configuration
19R-Acetoxytabersonine (1) : C23H26N2O4, colorless powder.1H NMR(600 MHz, CDCl3)δ: 8.99(1H, s, H-1), 7.22(1H, d,J=7.4 Hz, H-9), 7.15(1H, td,J=7.7, 1.0 Hz, H-11), 6.87(1H, td,J=7.4, 1.0 Hz, H-10), 6.83(1H, d,J=7.7 Hz, H-12), 5.94(1H, ddd,J=10.0, 5.0, 1.5 Hz, H-14), 5.77(1H, ddd,J=10.0, 2.5, 1.5 Hz, H-15), 4.56(1H, q,J=6.4 Hz, H-19), 3.73(3H, s, CO2CH3), 3.48(1H, ddd,J=16.0, 5.0, 1.5 Hz, H-3), 3.22(1H, dt,J=16.0, 2.0 Hz, H-3), 3.05(1H, ddd,J=8.3, 6.5, 1.5 Hz, H-5), 2.79(1H, s, H-21), 2.79(1H, m, H-17), 2.75(1H, m, H-5), 2.44(1H, d,J=15.3 Hz, H-17), 2.07(1H, m, H-6), 1.83(1H, m, H-6), 1.80(3H, s, CH3CO2), 0.92(3H, d,J=6.4 Hz, H-18);13C NMR(150 MHz, CDCl3)δ: 170.1(s, CH3CO2), 168.6(s,CO2CH3), 166.1(s, C-2), 143.5(s, C-13), 137.6(s, C-8), 129.8(d, C-15), 128.0(d, C-11), 127.0(d, C-14), 121.4(d, C-9), 120.8(d, C-10), 109.9(d, C-12), 91.8(s, C-16), 69.7(d, C-19), 66.7(d, C-21), 55.6(s, C-7), 51.1(q, CO2CH3), 51.0(t, C-5), 50.3(t, C-3), 45.9(s, C-20), 44.4(t, C-6), 27.5(t, C-17), 20.9(q, CH3CO2), 15.5(q, C-18); HR-ESI-MSm/z: 395 [M+H]+.
11-Methoxy-19-hydroxytabersonine (2) :C22H26N2O4, colorless oil.1H NMR(600 MHz, CDCl3)δ: 8.87(1H, s, H-1), 7.13(1H, d,J=8.0 Hz, H-9), 6.42(1H, d,J=2.2 Hz, H-12), 6.40(1H, m, H-10), 5.91(1H, ddd,J=10.0, 5.0, 1.5 Hz, H-14), 5.79(1H, m, H-15), 3.79(3H, s, OCH3), 3.78(3H, s, CO2CH3), 3.45(1H, ddd,J=16.0, 5.0, 1.5 Hz, H-3), 3.34(1H, q,J=6.4 Hz, H-19), 3.21(1H, dt,J=16.0, 2.0 Hz, H-3), 3.04(1H, ddd,J=8.5, 6.7, 1.8 Hz, H-5), 2.86(1H, dd,J=15.5, 2.0 Hz, H-5), 2.73(1H, m, H-17), 2.72(1H, s, H-21), 2.47(1H, d,J=15.4 Hz, H-17), 2.08(1H, ddd,J=11.7, 10.3, 6.7 Hz, H-6), 1.82(1H, ddd,J=11.7, 5.0, 1.8 Hz, H-6), 0.88(3H, d,J=6.3 Hz, H-18);13C NMR(150 MHz, CDCl3)δ: 168.7(s, CO2CH3), 167.0(s, C-2), 160.2(s, C-11), 144.4(s, C-13), 130.2(s, C-8), 129.6(d, C-15), 126.5(d, C-14), 122.1(d, C-9), 105.5(d, C-10), 96.9(d, C-12), 91.7(s, C-16), 67.1(d, C-21), 66.9(d, C-19), 55.6(q, OCH3), 55.1(s, C-7), 51.5(q, CO2CH3), 51.1(t, C-5), 50.3(t, C-3), 46.6(s, C-20), 44.1(t, C-6), 27.6(t, C-17), 17.5(q, C-18); HR-ESI-MSm/z: 383 [M+H]+.
16β-Hydroxy-19R-vindolinine (3) : C21H24N2O3, colorless oil.1H NMR(600 MHz, CD3OD)δ: 7.20(1H, d,J=7.4 Hz, H-9), 7.01(1H, t,J=7.7Hz, H-11), 6.73(1H, t,J=7.4 Hz,H-10), 6.69(1H, d,J=7.7 Hz, H-12), 6.27(1H, dd,J=9.7, 3.4 Hz, H-15), 5.85(1H, m, H-14), 3.89(1H, dd,J=17.5, 5.0 Hz, H-3), 3.72(3H, s, CO2CH3), 3.45(1H, ddd,J=17.5, 3.5, 2.0 Hz,H-3), 3.30(1H, s, H-21), 3.26(1H, d,J=9.2 Hz, H-5), 3.15(1H, m, H-5), 2.86(1H, dd,J=15.5, 2.3 Hz, H-17), 2.11(1H, m, H-19), 2.07(1H, m, H-6), 1.90(1H, d,J=15.5 Hz, H-17), 1.79(1H, ddd,J=15.7, 11.6, 8.8 Hz, H-6), 1.20(3H, d,J=7.2 Hz, H-18);13C NMR(150 MHz, CD3OD)δ: 174.0(s, CO2CH3), 150.8(s, C-13), 138.0(s, C-8), 132.3(d, C-15), 129.0(d, C-11), 128.6(d, C-14), 124.4(d, C-9), 120.7(d, C-10), 111.5(d, C-12), 84.8(s, C-16), 78.4(s, C-2), 78.0(d, C-21), 60.9(s, C-7), 58.6(t, C-5), 52.6(q, CO2CH3), 50.8(t, C-3), 49.8(d, C-19), 47.5(s, C-20), 40.7(t, C-17), 36.9(t, C-6), 10.1(q, C-18); HR-ESI-MSm/z: 353 [M+H]+.
19R-Kitramine (4) : C22H26N2O4, colorless oil.1H NMR(600 MHz, CDCl3)δ: 7.09(1H, dd,J=7.5, 1.2 Hz, H-9), 7.02(1H, td,J=7.6, 1.2 Hz, H-11), 6.67(1H, td,J=7.4, 1.1 Hz, H-10), 6.63(1H, dd,J=7.8, 1.1 Hz, H-12), 5.89(1H, ddd,J=9.8, 4.9, 1.6 Hz, H-14), 5.36(1H, dt,J=9.8, 2.1 Hz, H-15), 3.86(1H, q,J=6.5 Hz, H-19), 3.81(3H, s, CO2CH3), 3.52(1H, ddd,J=16.2, 4.9, 1.6 Hz, H-3), 3.27(1H, m, H-5), 3.22(3H, s, OCH3), 2.93(1H, dt,J=16.2, 2.1 Hz, H-3), 2.68(1H, m, H-6), 2.65(1H, m, H-5), 2.61(1H, s, H-21), 2.53(1H, d,J=12.5 Hz, H-17), 2.29(1H, dd,J=12.5, 2.0 Hz, H-17), 1.73(1H, m, H-6), 0.56(3H, d,J=6.5 Hz, H-18);13C NMR(150 MHz, CDCl3)δ: 171.9(s, CO2CH3), 148.3(s, C-13), 137.8(s, C-8), 128.8(d, C-14), 127.7(d, C-11), 127.5(d, C-15), 122.6(d, C-9), 118.4(d, C-10), 108.3(d, C-12), 100.0(s, C-2), 85.9(s, C-16), 82.7(d, C-19), 72.6(d, C-21), 56.8(s, C-7), 54.3(t, C-5), 52.6(t, C-3), 52.6(q, CO2CH3), 51.6(q, OCH3), 48.7(s, C-20), 38.1(t, C-17), 36.9(t, C-6), 14.0(q, C-18); HR-ESI-MSm/z: 383 [M+H]+.
Trichophylline (5) : C21H22N2O4, colorless crystal(MeOH).1H NMR(600 MHz, CDCl3)δ: 10.65(1H, s, H-1), 9.56(1H, d,J=1.6 Hz, H-19), 7.19(1H, t,J=7.8 Hz, H-11), 7.12(1H, d,J=7.5 Hz, H-9), 6.93(1H, t,J=7.5Hz, H-10), 6.84(1H, d,J=7.8 Hz, H-12), 5.68(1H, dt,J=12.4, 1.9 Hz, H-15), 5.63(1H, m, H-14), 4.82(1H, dt,J=15.6, 3.0 Hz, H-3), 3.78(3H, s, CO2CH3), 3.64(2H, m, H-5), 3.29(1H, dd,J=15.7, 7.2 Hz, H-3), 2.91(1H, d,J=15.6 Hz, H-17), 2.66(2H, td,J=9.0, 5.3 Hz, H-6), 2.45(1H, d,J=15.6 Hz, H-17), 1.33(3H, s, H-18);13C NMR(150 MHz, CDCl3)δ: 203.1(s, C-19), 170.8(s, CO2CH3), 170.4(s, C-21), 165.5(s, C-2), 144.2(s, C-13), 141.7(d, C-15), 132.6(s, C-8), 129.2(d, C-11), 123.6(d, C-14), 122.7(d, C-9), 121.8(d, C-10), 109.6(d, C-12), 90.3(s, C-16), 60.5(s, C-7), 51.6(s, C-20), 51.2(q, CO2CH3), 44.6(t, C-5), 40.6(t, C-3), 33.9(t, C-17), 31.2(t, C-6), 29.4(q, C-18); HR-ESI-MSm/z: 383 [M+H]+.
Voaharine (6) :C19H22N2O3, yellow crystal(MeOH).1H NMR(600 MHz, CD3OD)δ: 7.56(1H, d,J=7.6 Hz, H-12), 7.33(1H, m, H-9), 7.32(1H, m, H-10), 7.25(1H, t,J=7.6 Hz, H-11), 3.58(1H, d,J=13.5 Hz,H-17), 3.24(1H, m, H-14), 3.12(1H, m, H-15), 3.11(1H, m, H-3), 2.79(1H, d,J=12.0 Hz, H-21), 2.66(1H, dd,J=13.5, 2.3 Hz, H-3), 2.61(1H, td,J=14.7, 13.4, 4.8 Hz, H-5), 2.40(1H, d,J=13.5 Hz, H-17), 2.27(1H, m, H-5), 2.22(1H, m, H-6), 2.02(1H, d,J=12.0 Hz, H-21), 1.98(1H, m, H-6), 1.67(2H, q,J=7.4 Hz, H-19), 1.10(3H, t,J=7.4 Hz, H-18);13C NMR(150 MHz, CD3OD)δ: 191.5(s, C-16), 172.5(s, C-2), 141.6(s, C-13), 140.8(s, C-8), 129.7(d, C-10), 129.0(d, C-11), 128.3(d, C-9), 125.7(d, C-12), 77.4(s, C-7), 59.4(d, C-15), 55.8(t, C-21), 53.7(d, C-14), 52.3(t, C-3), 51.0(t, C-5), 44.8(t, C-6), 40.3(s, C-20), 38.8(t, C-17), 33.5(t, C-19), 8.0(q, C-18); HR-ESI-MSm/z: 327 [M+H]+.
X-ray crystallographic data for voaharine (6) : A light yellow BLOCK-like of C19H22N2O3,M=326.38, approximate dimensions 0.229 mm x 0.246 mm x 0.384 mm, was used for the X-ray crystallographic analysis. The integration of the data using a orthorhombic unit cell yielded a total of 20084 reflections to a maximum θ angle of 79.34°(0.78 Å resolution), of which 3506 were independent(average redundancy 5.728, completeness=98.4%,Rint=4.16%,Rsig=3.46%) and 3467(98.89%) were greater than 2σ(F2). The final cell constants ofa=6.5144(4) Å,b=8.7698(6) Å,c=28.806(2) Å,α=90.00°,β=90.00°,γ=90.00°,V=1645.69(19) Å3,T=104(2) K. Data were corrected for absorption effects using the Multi-Scan method(SADABS). The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group C121,Z=2,μ(CuKα)=1.54178. The final anisotropic full-matrix least-squares refinement on F2with 222 variables converged atR1=3.61%, for the observed data andwR2=10.11% for all data. The goodness-of-fit was 1.079. The absolute configuration was determined by the Flack parameter=0.06(5), which was determined using 1424 quotients [(I+)-(I-)]/[(I+)+(I-)].The crystallographic data for6was deposited in the Cambridge Crystallographic Data Centre(CCDC deposition numbers: 1985749).
The cytotoxicity assay results showed that1and2exhibited good cytotoxic effects against five human cancer cell lines with IC50values comparable to that of cisplatin(Tab. 1).
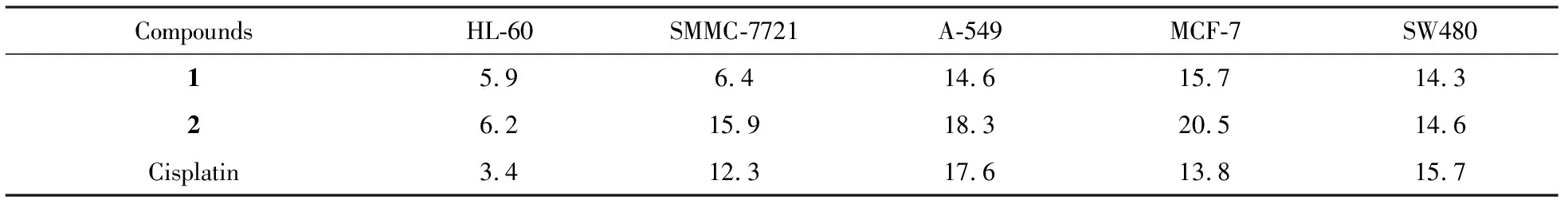
Tab.1 Cytotoxicity of compounds 1 and 2 (IC50, μM)
3 Conclusion
Six alkaloids were isolated fromM.henryiand identified as 19R-acetoxytabersonine(1),11-methoxy-19-hydroxytabersonine(2),16β-hydroxy-19R-vindolinine(3),19R-kitramine(4),trichophylline(5),and voaharine(6). Compounds1and2displayed significant cytotoxicity against five human cancer cell lines. This study laid a foundation for the following research on the chemical constituents and their biological activities ofM.henryi.
猜你喜欢
Zoological Research(2022年5期)2022-10-17
中国典型病例大全(2022年10期)2022-05-10
广西医科大学学报(2021年12期)2022-01-24
中山大学学报(自然科学版)(中英文)(2021年3期)2021-05-26
中国应急管理科学(2021年9期)2021-03-16
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
植物研究(2020年6期)2020-03-05
时代风采(2019年4期)2019-12-14
药学实践杂志(2011年6期)2011-11-22
