
N-氯乙酰水杨酰肼镍(Ⅱ)配合物的合成、表征和水解活性(英文)
2020-08-03 00:38金龙飞王梦痕冯晓红张巧元吴腊梅
中南民族大学学报(自然科学版) 2020年4期
金龙飞,王梦痕,冯晓红,张巧元,吴腊梅
(中南民族大学 化学与材料科学学院,武汉 430074)
Urease (urea amidohydrolase; E.C.3.5.1.5) is a nickel-containing enzyme present in various bacteria,fungi,and plants where it catalyzes the urea degradation to supply nitrogen source for the growth of these organisms. The enzyme has also been found to be important in the virulence ofHelicobacterpyloriandProteusmirabilis. Besides,urease also plays an important role in seed germination by degrading urea formed from arginine. The chemistry nature of urease is identified as catalyzing enzyme which can catalyze the hydrolysis of urea to form ammonia and carbamate,which spontaneously decomposes into carbonic acid and a second ammonia molecule[1].
Several crystal structures of the wild-typeurease have been published,which were found to be metalloproteins with three dinuclear nickel sites,with the two nickel ions in each site separated by about 3.5 Å[2-3]. Each nickel ion is in the native structure coordinated by two histidine residues and one water molecule,while a carbamylated lysine residue and one hydroxyl group bridge the two ions. A number of crystal structures ofKlebsiellaaerogenesandBacilluspaseuriiurease with bound inhibitors have also been published[4].
Some enzymatic mechanisms of urease have been proposed[5]. A common feature in the published mechanistic proposals is that the reaction is initiated by the coordination of the urea oxygen to the coordinatively unsaturated nickel ion. The source of the attacking hydroxide has been under debate[6]. Both the deprotonated terminal water molecule on the second Ni2+and the bridging hydroxide have been suggested.
To study the proposed mechanisms,we attempted to prepare a structural and functional mimic of the urease active site. Here we reported the synthesis of the ligandN-chloroacetyl-salicylhydrazide (H3cashz) (3) (Fig.1),and the preparation of trinuclear nickel complex4(Fig.1). The hydrolytic activity of the complex was tested in the hydrolysis of the organophosphoester 2-hydroxypropylp-nitrophenyl phosphate (HPNPP)[7].

Fig.1 Synthetic route and chemical structures of the compound 1,2,3,4
1 Experimental
1.1 Materials
All chemicals and solvents were reagent grade or biochemical reagent and used as purchased. Methanol,ethanol andN,N-dimethylformamide (DMF) were used without purification. Methyl salicylate,hydrazine hydrate,chloroacetic acid,thionyl chloride,nickel acetate tetrahydrate,sodium perchlorate,sodium hydrate,ethanol,methanol,DMF,chloroform,acetonitrile and pyridine were purchased from China Sinopharm Group Chemical Reagent Co.,Ltd.,Shanghai,China. Salicylhydrazide (2) was prepared according to literature procedures[8]. Chloroacetyl chloride as an intermediate was prepared according to literature procedure[9].
1.2 Characterization
Infrared spectra were measured on a Thermo Nicolet Corporation NEXUS FT-IR spectrometer as KBr pellets in the 4000-400 cm-1region. UV-vis spectra were recorded on a Shimadzu-UV-2501 PC spectrophotometer.1H NMR and13C NMR spectra were recorded on a Varian Inova 600 MHz NMR spectrometer at 25 ℃. Chemical shifts were referenced to residual solvent peak. C,H and N elemental analysis were performed on a Perkin Elmer 2400 CHN elemental analytical instrument,Ni was determined by atomic absorption spectroscopy on a Perkin Elmer 1100B spectrophotometer. Single crystal X-ray diffraction was performed on a Bruker Smart APEX diffractometer,and intensity data were collected with a graphite monochromatic MoKa radiation (λ=0.71073 Å) at 292(2) K.
1.3 Synthesis of N-chloroacetyl salicylhydrazide (H3cashz) (3)
The ligand H3cashz was synthesized by reacting chloroacetyl chloride (11.30 g,0.10 mol) with salicylhydrazide (7.61 g,0.05 mol) in 200 mL of chloroform at 0 ℃. The reaction mixture was stirred for 1 h,then heated to boiling point and stirred for 5 h. After the mixture was concentrated under reduced pressure,the residue was filtered and rinsed with water. White solid was obtained by recrystallization with water and alcohol. Yield: 8.93 g,78%. m.p. 223-225 ℃. Found: C 47.21,H 3.89,N 12.21; calc. for C9H9ClN2O3: C 47.28,H 3.97,N 12.25. IR (KBr pellet,cm-1):υO-H3190 s,broad;υN-H3266 s,broad;υC=O1655 vs;υC=N-C=N1608 s.1H NMR (DMSO-d6)δ: 11.80 (s,1H,-Ph-OH),10.70 (s,1H,-NH-CO-CH2Cl),10.51 (s,1H,-Ph-CO-NH-),7.87 (d,1H,o-PhCH),7.49 (m,1H,p-PhCH),6.98-6.96 (m,2H,m-PhCH),4.23 (s,2H,-CH2Cl).13C NMR (DMSO-d6)δ: 166.73 (-CO-CH2Cl),165.04 (-Ph-CO-),159.18 (-PhC-OH),134.58 (p-PhC),129.08 (o-PhC),119.61 (m-PhC),117.74 (-PhC-CO-),115.16 (m-PhC-Ph-OH),41.28 (-CH2Cl).
1.4 Synthesis of nickel complex
([Ni3(C9H6N2O3Cl)2(C3H7NO)2(C5H5N)2]) (4)
H3cashz (0.23 g,1.0 mmol) was dissolved in 30 mL of methanol,and 0.24 g (1.0 mmol) of nickel acetate tetrahydrate was dissolved in 10 mL of methanol in another flask. The two solutions were mixed and stirred for 1 h,then the solution was adjusted to pH of 4-5 with 1.0 mol·L-1pyridine methanol solution. After slow evaporation of the mother liquor in three days,red block crystals were obtained from the filtrate. Yield: 0.32 g,68%. Found: C 43.74,H 3.80,N 11.92,Ni 18.88; calc. for C34H36Cl2N8Ni3O8: C 43.83,H 3.89,N 12.03,Ni 18.90. IR (KBr pellet,cm-1):υC=O1652 vs,υC=N-C=N1604 s,υNi-N570 s. UV-Vis (CH3OH,nm),λmax: 332,205.
1.5 X-Ray analysis
Crystals of4suitable for X-ray were obtained as described above. A crystal of4with dimensions of 0.20 mm×0.10 mm×0.10 mm was mounted in a glass capillary with the mother liquor during X-ray diffraction data collection. Intensity data were collected with a graphite monochromatic MoKaradiation (λ=0.71073 Å) at 292(2) K on a Bruker Smart APEX diffractometer. From a total of 11129 reflections corrected by multi-scan absorption correction of XPREP[10-11]in the 2.07≤θ≤25.00 range,3391 were independent,of which 2234 observed reflections withI> 2σ(I) were used in the structural analysis. The structure was solved by direct methods. All non-hydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atoms were located in calculated positions. The positions and anisotropic thermal parameters of all non-hydrogen atoms were refined onF2by full-matrix least-squares techniques with SHELXTL program package[10-11]. The final refinement converged atR1=0.0544,wR2=0.0919,w=1/[σ2(Fo2)+(0.0286P)2+0.0000P],whereP=(Fo2+2Fc2)/3,(Δ/σ)max= 0.016,S= 0.916,(Δρ)max=0.449 and (Δρ)min= -0.371 e/Å3.
1.6 Hydrolytic activity
Hydrolytic activity was performed in 3 mL UV cells in 1∶1 H2O/MeCN (V∶V) solution. The solution was buffered using HEPES buffers (0.01 mol·L-1total concentrated). The ionic strength was maintained at 0.1 by addition of sodium perchlorate (0.1 mol·L-1). Dissolved complexes was added to a total concentration of 0.25 mmol of trinuclear complex,and HPNPP (0.82 mmol/L) was used as substrate. All the ingredients were mixed in a thermostatic cell (25 ℃),and the visual spectrum was recorded at 400 nm,where the extinction coefficient for the hydrolysis productp-nitrophenolate is 18500 L·mol-1·cm-1. The data were plotted,and the linear slope was used to deduce the initial rates. The total amount ofp-nitrophenol/phenolate was determined by using the pKa (7.15)[12]. The experiment was reproduced at five different pH between 7 and 9 (the optimum pH of urease is 8).
2 Results and discussion
2.1 Synthesis of the complex
Nickel complex4was synthesized by Ni(CH3COO)2·4H2O and deprotonatedN-chloroacetyl-salicylhydrazide (H3cashz) in methanol (Fig.1,followed by the addition of the 1.0 mol·L-1pyridine methanol solution. Along with the reaction,the color of the mixture was changed to red. The final complex is red crystalline solids,soluble in methanol and ethanol.
2.2 Spectral characterization
In the IR spectra,the ligand3shows stretching bands attributed to CO,CN,and NH at 1655,1608,and 3266 cm-1,respectively. Band at 3190 cm-1is assigned toυO-Hvibration which may involve intramolecular hydrogen bonding. In addition,a strong band found at 1608 cm-1is assigned to the CN-NC group.
In the complex4,the absence of the N-H stretching vibration band and weakened CO stretching vibration band is consistent with the deprotonation of the CONH groups and coordination to the Ni2+ion. This is also confirmed by the band at about 570 cm-1assigned to Ni-N. The CN-NC framework seen at l608 cm-1in the ligand shifted to 1604 cm-1upon coordination to Ni2+ion. The disappearance of the band at 3190 cm-1also supports the involvement of phenolic oxygen in coordination through deprotonation.
The1H NMR of the ligand showed a singlet atδ4.23 for the methyl chloride protons,a multiplet atδ6.96-6.98 for the aromatic protonsm-ArH,a multiplet atδ7.45 for the aromatic protonp-ArH,a singlet atδ7.87 for the aromatic protono-ArH,two singlet atδ10.51 and 10.70 for the hydrazine protons,a singlet atδ11.80 for the phenolic hydroxyl proton. In the13C NMR spectra of the ligand,four peaks at 115,120,129 and 135 are attributed to the four isolated carbons of aromatic ring,in addition,the other two carbons at 118 are assigned to the carbon linked with CO group,and 159 to the carbon linked with OH group. The methyl chloride carbon at 41 falls into the 40-42,the two carbonyl carbons are present in the 165-167 region. All these assignments are in good agreement with the spectra obtained.
2.3 Crystal structure of 4
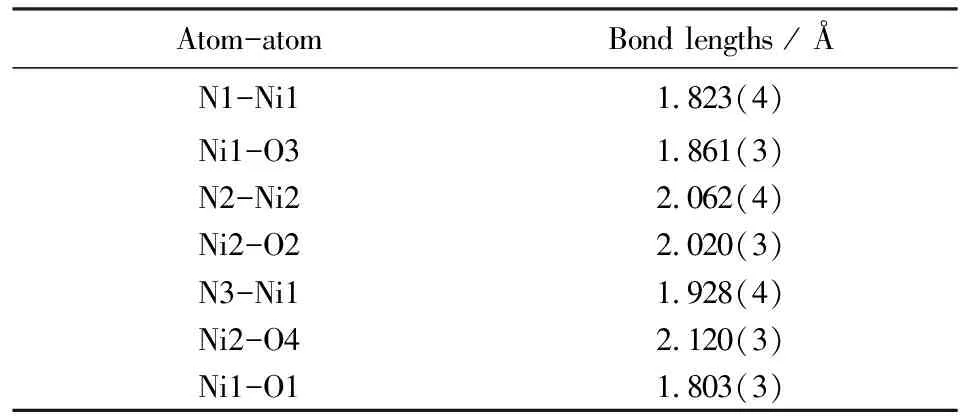
The complex4crystallizes in the monoclinic system and space groupP2(1)/n. A diagram of the crystal structure of complex4was presented in Fig.2. The crystallographic data were summarized in Tab.1,and selected bond distances and angles were given in Tab.2 and Tab.3.

Fig.2 Perspective view of the complex 4

Tab.1 Crystallographic data
Tab.2 Selected bond lengths in complex 4

Tab.3 Selected bond angles in complex 4
The structure of complex4exhibits a trinuclear arrangement of nickel ions linked by two deprotonated ligands cashz3-. The deprotonated ligand cashz3-acts as a trianionic pentadentate ligand,one phenolate oxygen,one carbonyl oxygen and one hydrazide nitrogen in the ligand are bound to the Ni2+cation at the side,and the other carbonyl oxygen plus the other hydrazide nitrogen in the same ligand are chelated to the center Ni2+cation. The two side nickel ions are of the same coordination environment with square planar geometry. The nickel ion is bonded by two nitrogen atoms (N1,N3) and two oxygen atoms (O1,O3) with one nitrogen atom (N3) from the second ligand pyridine and the other three coordinated atoms from cashz3-,one phenolate oxygen (O1),one carbonyl oxygen (O3) and one hydrazine nitrogen atom (N1). The bond length Ni1-N3 [1.928(4) Å] is longer than the other bond lengths [Ni1-N1 1.823(4) Å,Ni1-O1 1.803(3) Å,Ni1-O3 1.861(3) Å],which may be contributed to the stronger chelation of the backbone ligand. Deviation of Ni1 is only 0.0020 Å from the Ni1,N1,N3,O1,O3 plane,so the overall geometry around the four-coordinate nickel ion can be described as a square plane with r.m.s deviations of 0.0088 Å. The angles N2-Ni2-N2(A) and O2-Ni2-O2(A) are 180°,so the five atoms [Ni2,N2,N2(A),O2 and O2(A)] are in the same-plane. The angle O4-Ni2-O4A is 180° too,and the bond lengths of Ni2 with the coordinated atoms is close to the slightly longer bond Ni2-O4 [2.120(3) Å]. Thus the central nickel ion adopts an ideal six-coordinate octahedral geometry. The three nickel ions are in a line,and the distance between Ni1 and Ni2 is 4.612 Å. In addition,it can be seen from Fig.3 that the axially coordinated DMF molecules are disordered.

Fig.3 Concentration-absorbance curves of complex 4 in methanol measured at 205 nm(a)and 332 nm(b)
2.4 Structural stability of complex 4 in methanol
To explore the stability of complex in methanol solution,the concentration-dependent absorbance was measured at 205 nm and 332 nm for4,respectively (Fig.3). The absorbance increases linearly with the concentration at the range of 3.5 and 54.8 μmol·L-1. No change was observed for the solutions′ absorption after 24 h,indicating that complex4retain integrity of the trinuclear structure and is stable in methanol.
2.5 Reactivity study of compound 4
The catalytic activity of compound4was studied for the hydrolysis of 2-hydroxypropylp-nitrophenyl phosphate (HPNPP)[12-13]. In the assay,a water/MeCN solution (volume ratio of 1∶1) containing buffer,catalyst,HPNPP,and sodium perchlorate was prepared and the evolution ofp-nitrophenolate at 400 nm was studied. The result was then converted to nitrophenolate concentration and calculated to totalp-nitrophenol/p-nitrophenolate concentration by using the pKa for the acid/base pair. As can be seen in Fig.4,the reaction is faster at higher pH 8-9 for both the catalyzed and the uncatalyzed reaction,which may be expected since hydroxyl groups are more potent nucleophiles than water[5]. The activity of compound4is approximately one order of magnitude higher.

Fig.4 Initial hydrolysis rates curves of HPNPP at different pH values
3 Conclusion
In this work,a trinuclear nickel complex4was synthesized via the reaction of Ni(CH3COO)2·4H2O with the deprotonatedN-chloroacetyl-salicylhydrazide (H3cashz). The crystal structure of complex4showed that three Ni2+ions in a linear arrangement were bridged by the ligand cashz3-,with two four-coordinate square-planar nickel ions linked by a six-coordinate octahedral central nickel ion. This novel trinuclear nickel complex showed high catalytic capacity for the hydrolysis of 2-hydroxypropylp-nitrophenyl phosphate (HPNPP) at basic pH condition.
Supplementarymaterial
CCDC 1406749 contains the supplementary crystallographic data for complex4. The data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre,12 Union Road,Cambridge CB21EZ,UK; fax: +44 1223 336 033; or deposit@ccdc.cam.ac.uk).
猜你喜欢
政工学刊(2022年6期)2022-05-27
纺织科学研究(2021年6期)2021-12-02
大学生(2021年9期)2021-09-28
——材料科学与工程
湖南人文科技学院学报(2020年3期)2020-06-08
作文评点报·低幼版(2020年16期)2020-04-20
中南医学科学杂志(2019年4期)2019-08-12
艺术评论(2018年3期)2018-05-16
金山(2016年10期)2016-11-25
海峡科技与产业(2016年3期)2016-05-17
创新科技(2015年4期)2015-12-24
