
大粒径氧化石墨及石墨烯的制备与表征
2020-07-23 04:47:26常然庞秀言李泽江信亚平
河北大学学报(自然科学版) 2020年3期
常然,庞秀言,2,李泽江,信亚平
(1.河北大学 化学与环境科学学院,河北 保定 071002;2.河北省阻燃材料工程技术研究中心,河北 保定 071002)
氧化石墨(GO)及其还原产物石墨烯以其独特的物理、化学特性,受到了物理、化学、电子、信息技术、材料、能源及生物等领域的广泛关注[1-2].石墨烯的主要制备方法有微机械剥离法[3]、外延生长法[4]、石墨插层法[5]、溶液剥离法[6]、化学气相沉积法[7]以及氧化还原法[8-10]. 以石墨的氧化反应为合成路线的制备方法是用强氧化剂对石墨进行氧化处理,首先得到在石墨表面键合-OH、环氧基、-COOH等含氧基团的GO中间产物,再经超声、还原得到石墨烯. 由于化学氧化法反应条件易于控制、成本相对较低,而在石墨烯制备中得到了广泛应用. 化学氧化法又以Hummers法应用最为广泛[11]. 然而,Hummers法的主要缺点表现为低温氧化与高温水解时间短,氧化插层反应不彻底. 改进的Hummers法通过延长低温与高温反应时间使得后期的氧化、插层、水解膨胀更加充分[12-13],得到的GO的堆叠层数降低.
不同尺寸、结构的GO和石墨烯性能存在差异. 研究表明[14]添加大粒径石墨烯的环氧树脂复合材料的热导率高于小粒径石墨烯改性材料.用粒径0.045 mm石墨粉制备的GO比0.001 3 mm石墨粉制备的GO对材料的热稳定性、玻璃化温度影响更加显著[15].相比于大粒径石墨,小粒径石墨存在更多的结构缺陷. 因而,氧化剂更容易插入石墨片层之间, 从而使得GO的层间距变大,容易剥离. Qian等[16]采用KMnO4、K2S2O8、H2SO4、P2O5为氧化剂,借助于控制预氧化反应温度、去离子水稀释浓度获得了1~7 μm的GO.而采用电化学法得到3 μm的石墨烯的产率可以达到8.7%[17].Zhang等[18]制备了表面键合大量-OH基团,尺寸为1.5 μm的GO.大粒径石墨晶格缺陷相对较少,因而氧化、插层困难.文献中未见有以0.10~0.30 mm石墨为原料制备粒径10 μm以上的GO和石墨烯的报道.本研究在改进的Hummers法基础上,采用密闭氧化法[19],以0.10~0.30 mm鳞片石墨为原料,浓H2SO4、KMnO4为氧化剂,在冷反应中通过延长反应时间使原料得以充分预氧化,为其在高温下充分氧化、剥离而得到大粒径GO提供了保障.通过正交实验、单因素实验优化了GO、石墨烯的制备条件,并借助于扫描电镜、X线衍射、傅里叶红外光谱、原子力显微镜、热重、拉曼光谱等手段对产物的形貌、结构、热稳定性及组成进行了表征.
1 实验部分
1.1 仪器与试剂
TDZ5M台式低速自动平衡离心机(湖南省长沙市易达仪器);DHG-9075A型电热鼓风干燥箱(上海飞跃);D8 ADVANCE型X线衍射仪(德国BRUKER-AXS有限公司);TM3000台式扫描电子显微镜(SEM)(日本株式会社日立高新技术公司);TENSOR27傅里叶变换红外光谱仪(FTIR)(布鲁克光谱仪器公司);Raman光谱仪LabRAM HR Evolution(日本,HORIBA);STA449C-QMS403C型热失重分析仪(TG)(德国NETZSCH公司);5500AFM/SPM原子力显微镜(AFM)(美国安捷伦公司).
0.10、0.15、0.18、0.30 mm石墨(碳质量分数为96%)由青岛西特碳素有限公司提供,KMnO4、浓H2SO4(质量分数98%)、氨水、H2O2、盐酸、四氢呋喃为市售分析纯试剂.
1.2 GO的制备
将水热合成反应釜以及按剂量称量的石墨、浓H2SO4、KMnO4在冰浴中冷却,然后将反应物一并转移至反应釜中,借助工具立即旋紧盖子,置于冰浴中进行冷反应.冷反应完成后,将反应釜转移至烘箱中,在80 ℃下进行热反应.热反应完成后,反应釜冷却到室温,将产物转移至烧杯,先用1.0 L去离子水进行稀释,再加入适量的H2O2,不断搅拌,溶液颜色转为黄褐色后进行过滤,滤掉上层固体.将收集的滤液分别用质量分数5%HCl、去离子水进行洗涤并离心,直至pH达到5.0左右,取出底层物,在50 ℃下烘干24.0 h得到GO.
冷反应与热反应时间、浓H2SO4及KMnO4用量均为影响GO质量与产率的因素. 实验中分别以2.0 g0.10、0.15、0.18、0.30 mm的4种不同粒径石墨为原料,通过正交实验优化了冷反应时间、热反应时间、浓H2SO4及KMnO4用量,4种因素的取值见表1. 以所得GO产率高且纯度好为目标,筛选、确定各粒径石墨制备GO的条件.

表1 GO制备正交实验影响因素及其水平a
1.3 石墨烯制备
依照氨-水合肼法[20-21],将1.0 g的GO在超声、机械搅拌下分散于去离子水中,期间加入5.0 mL的氨水,1.0 h后加2.0 mL的水合肼,2.0 h后升温至99 ℃继续还原反应24.0 h,反应完毕,反应体系降至室温,洗涤至pH为7.0左右,在50 ℃下烘干24.0 h,制得石墨烯.
1.4 样品表征
1.4.1 化学结构分析
采用FTIR分别对制备的GO、石墨烯进行化学结构分析.将样品与溴化钾按照1∶100的质量比混合、研磨,于20 MPa下压片后,以2 cm-1扫描频率在4 000~400 cm-1内测试.
1.4.2 物理结构测定
测试过程采用514.5 nm的氩激光激发样品,分别测定GO、石墨烯的拉曼光谱(Raman).
XRD分析:采用Ni过滤的Cu Kα射线,管电压为40 kV,管电流为40 mA下,以4°/min的速率连续扫描GO、石墨烯样品,通过XRD分析确定原子的结构形态.
1.4.3 形貌分析
借助TM 3000台式SEM分别对GO、石墨烯外观形貌实施测定.
石墨烯经研磨、超声分散于乙醇溶剂中,然后均匀滴加分散于云母片.经烘干后用AFM观察样品厚度.
1.4.4 热重分析
在N2气氛的30 mL/min流量下,将约10 mg样品分别放置于事先固定好的坩埚中,以10 ℃/min的升温速率从室温升至800 ℃,得GO、石墨烯的用以进行热稳定性分析的TG曲线.
2 结果与讨论
2.1 GO制备条件优化
冷反应时间、热反应时间、浓H2SO4及KMnO4用量是影响GO产率与纯度(以产品质量与原料石墨质量之比计算[22])的关键因素. 实验阶段,分别以2.0 g 的0.10、0.15、0.18、0.30 mm 4种不同粒径石墨为原料,根据表1,以所得产品纯度与产率为目标,采用L9(34)正交实验对表中各因素取值进行优化. 将制备的各GO分别记作GO1、GO2、GO3以及GO4.正交实验结果表明:以2.0 g石墨为原料基准,加入8.0 g KMnO4,70 mL 浓H2SO4,分别在冰浴条件下预氧化8.0 h,80 ℃下加热反应2.0 h,最终得到的GO产率高,纯度好.研究表明冷反应时间是制备高纯度及产率的GO1、GO2、GO3、GO4关键因素,并且,冷反应时间越大,各产品的产率越高.KMnO4及H2SO4用量、热反应时间对产率影响较小.
由于冷反应时间为影响GO产率的关键因素,进一步的单因素实验中以2.0 g的0.10 mm石墨做原料,加入70 mL浓H2SO4、8.0 g KMnO4,在4~14 h内改变冷反应时间,对石墨进行不同程度的预氧化,然后于80 ℃下继续反应2.0 h. 如表2所示,GO4产率随冷反应时间增加而提高,增至8.0 h后,冷反应时间对产率的影响明显变弱. 可以看出8.0 h后石墨的预氧化反应初步完成.
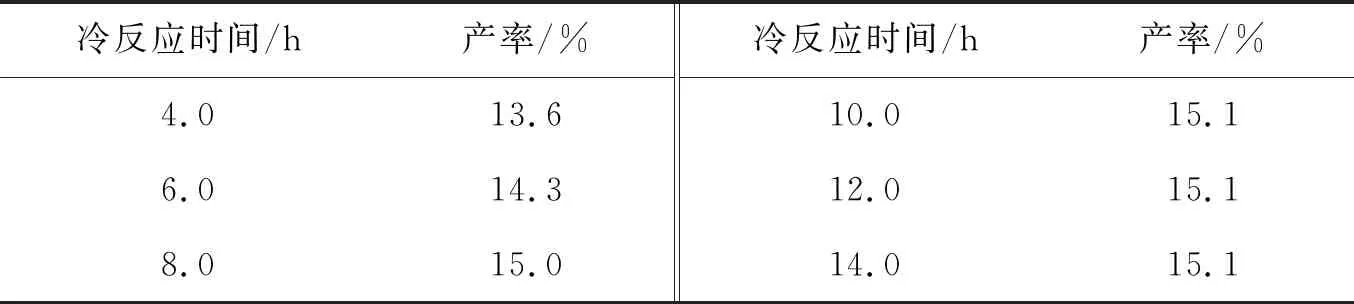
表2 预氧化反应时间与GO4产率的关系
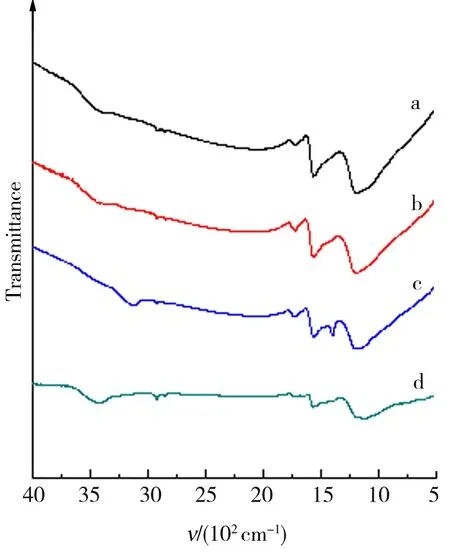
为进一步确定冷反应对GO的结构及反应程度的影响,分别对预氧化产物及原料石墨进行了XRD以及FTIR分析.如图1可知,3 430 cm-1为原料未处理前在C原子上键合的-OH的伸缩振动吸收峰,增加冷反应时间,该峰强度逐渐减弱.在低波数区域产生了-COOH伸缩振动吸收峰(3 130 cm-1),其强度随冷反应时间增加逐渐增大,并且,羰基(-C=O-)的伸缩振动双峰(1 700~1 400 cm-1)亦逐渐加强. 同时,出现了C-O基的较强吸收峰(1 250~1 000 cm-1). 据此可以推测,在冰浴控温以及KMnO4与浓H2SO4的联合强氧化条件下,可以完成石墨碳的初步氧化,其表面上可生成-C=O-、C-O及-COOH基团[19],且原料的氧化程度随低温冷反应时间的延长而增强.
图2所示的XRD结果表明,实验中所用石墨于26°产生了极强衍射峰,与其002晶面对应,证实其Z轴方向的晶形保持完好. 尽管经不同冷反应时间所得产物于26°附近保留了002晶面特征衍射峰,然而此衍射峰强度相对原料石墨已经显著减弱,峰的宽亦明显增大,且向小角度产生了迁移. 这是由于KMnO4与浓H2SO4对碳原子的初步氧化导致了石墨层间距离变大.石墨层状结构的完整程度遭到破坏,其Z轴方向同向排列晶面数在减少,即导致石墨片层产生了不同程度剥离. 尤其是,冷反应时间越长,此特征越明显. 由此可见,低温氧化对石墨的组成与结构变化影响显著,可以称为GO制备中石墨的预氧化阶段[16].
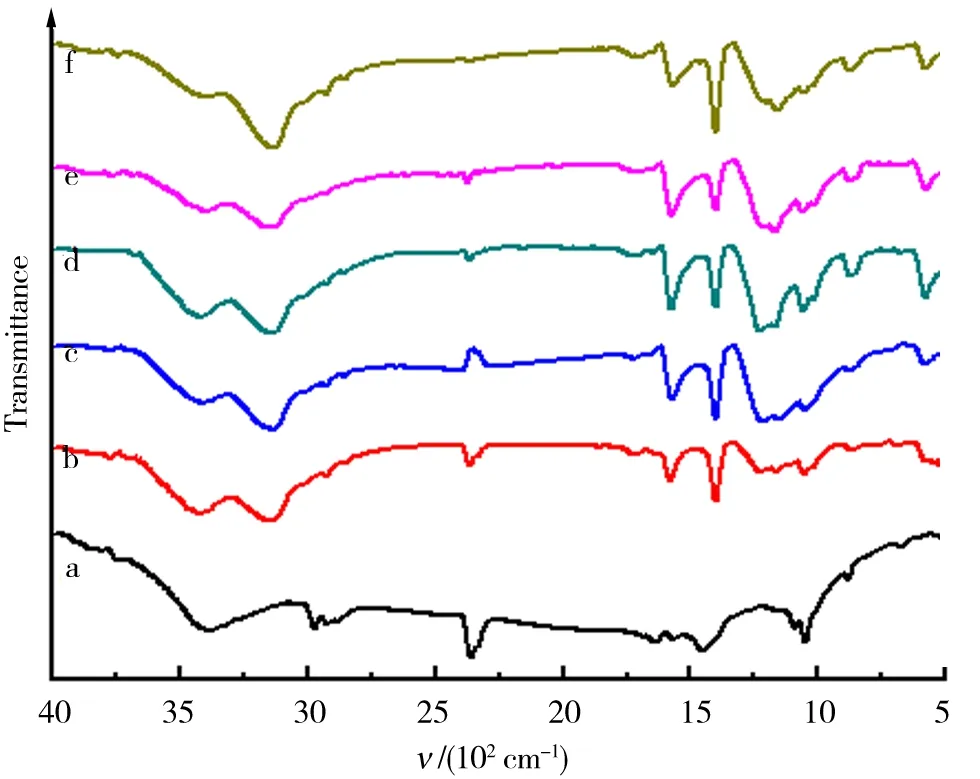
a—f.冷反应时间分别为0、2.0、4.0、6.0、8.0、10.0 h.图1 石墨与冷反应产物的FTIRFig.1 FTIR of graphite and theproducts from cold reaction
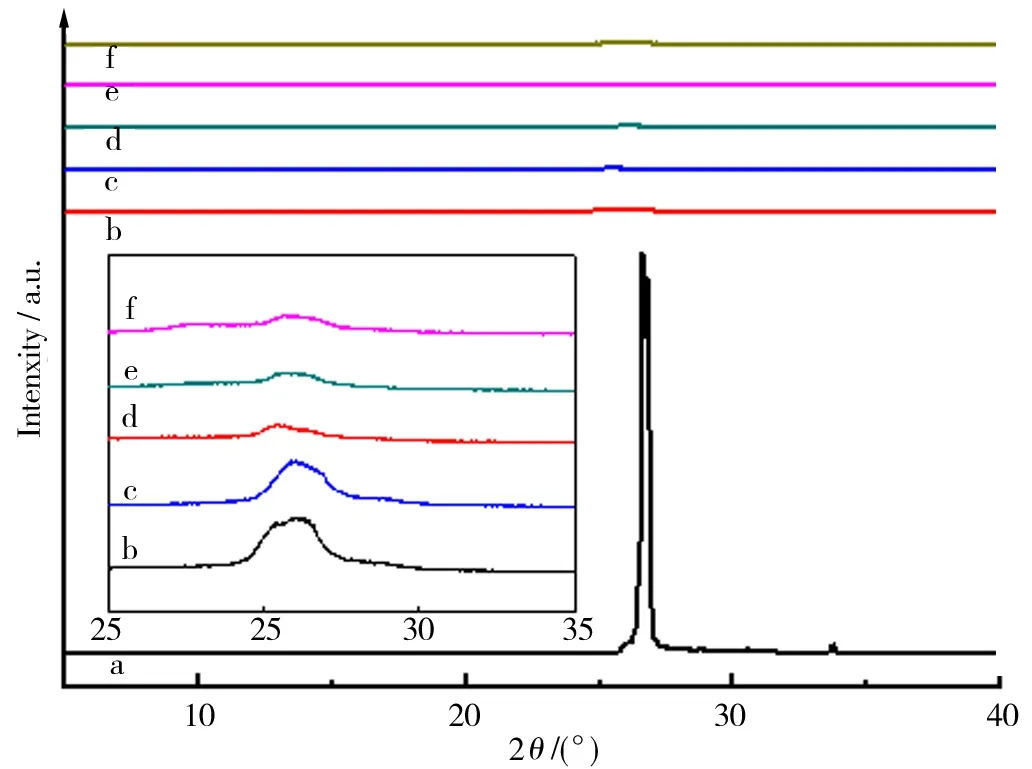
a—f.冷反应时间分别为0、2.0、4.0、6.0、8.0、10.0 h.图2 石墨与冷反应产物的XRDFig.2 XRD of graphite and the products from cold reaction
2.2 原料粒径对GO产率影响
按照1.2中步骤,各取4种粒径石墨2.0 g,在反应釜中依次加入经冰浴冷却的70 mL浓H2SO4、8.0 g的KMnO4,旋紧盖子.先置于冰浴中反应8.0 h,冷反应完成后,将反应釜转移至烘箱中,在80 ℃下继续热反应2.0 h,所得产物分别标记为GO1、GO2、GO3、GO4. 同时,为了与文献[22]对比,以0.075 mm石墨粉为原料,在上述实验条件下,进一步制备了GO5. GO产率以石墨质量为计算基准,结果见表3. 如表3所示,石墨粒径越小,GO的产率越大, 但产率增速是非线性的. 从GO1到GO3,产率增加1.0%;而从GO3到GO4,产率剧增11.4%,因而存在影响GO产率的临界点. 尤其是GO5的产率达到了103.0%. 此结果说明石墨粒径越小,其氧化越容易.
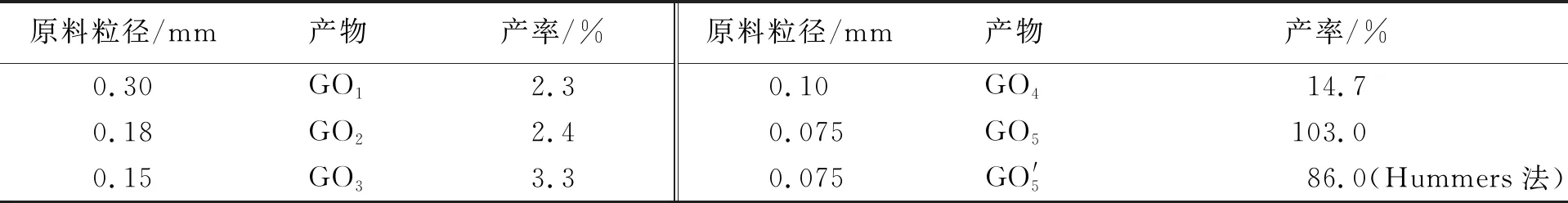
表3 原料粒径对GO产率的影响
进一步以石墨粉0.075 mm为原料,采用Hummers法制备了GO′5:经冰浴降温后,将360 mL的浓H2SO4加入到7.5 g石墨粉与7.5 g NaNO3的混合物中,缓慢加入45 g KMnO4,并控温在20 ℃以下,加热升温至35 ℃,在搅拌下反应3.0 h.将1.5 L的H2O2(质量分数3%)缓慢加入,98 ℃下控温、搅拌、继续反应0.5 h.反应完毕,在3 700 r/min下离心,取出上清液,剩余固相继续用去离子水洗涤.经多次洗涤、离心,洗液pH值接近7.0,剩余固体经干燥、称重,得GO′5的产率为86%.
将密闭氧化法与Hummers法对比可以看出:1)前者仅需冷反应和热反应2个阶段,而后者有冰浴控温和分别升温至35 ℃与98 ℃ 3个阶段;2)在密闭氧化法中,由于石墨的冷反应时间明显高于后者,使得KMnO4及浓H2SO4的用量也明显加大,但后者为提高氧化性需要另外添加NaNO3;3)相比而言,前者操作方便,且条件便于控制.Hummers法对反应条件要求高,操作相对危险,产生污染物也较多;4)密闭氧化法制备的GO5产率高于改进Hummers法.
2.3 GO与石墨烯的XRD结果
用XRD表征了制备所得GO与石墨烯的结构. 由图3可知各粒径石墨经过低温与高温2个阶段的氧化处理后,其在26°附近的002晶面对应衍射峰已经基本消失,但是在位于13.6°处新出现了宽而弱的衍射峰,对应晶面间距为0.649 nm. 说明氧化产物GO的层间距明显增加,氧化后石墨碳的晶格有序性已经严重破坏.其可能的原因在于C原子上引入了大量-OH、-C=O-、-COOH等官能团,使得石墨在Z轴方向间距增加, 并且呈现出碳的氧化程度随石墨粒径减小而不断增大的趋势.图4中显示,GO1、GO2、GO3、GO4的还原产物均没有产生明显特征峰,证实其均已剥离为石墨烯,Z轴方向的长程有序性已经消失. 然而,由于范德华力作用,干燥过程中石墨烯的部分单片层重新发生了堆叠而形成了多层石墨烯,再次产生长程有序性,因而在25°附近产生了微弱衍射[19].
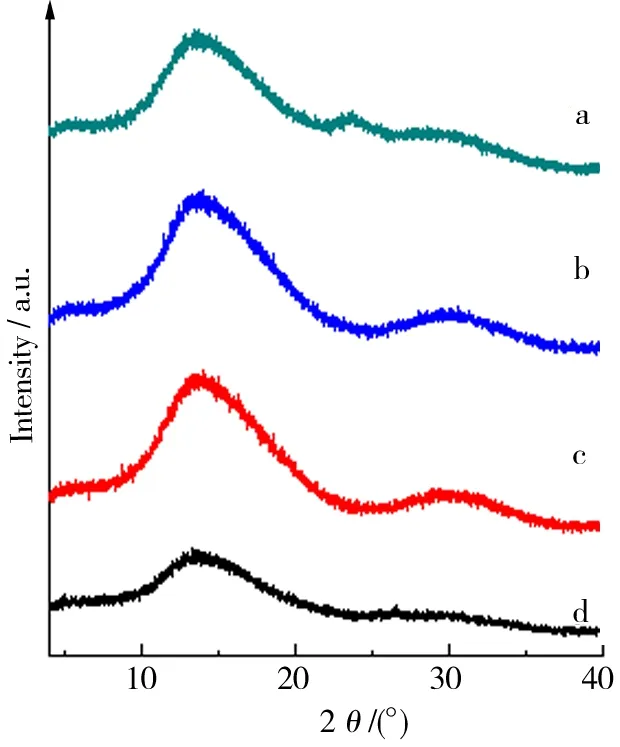
a.GO1;b.GO2;c.GO3;d.GO4.图3 GO的XRD结果Fig.3 XRD result of each GO
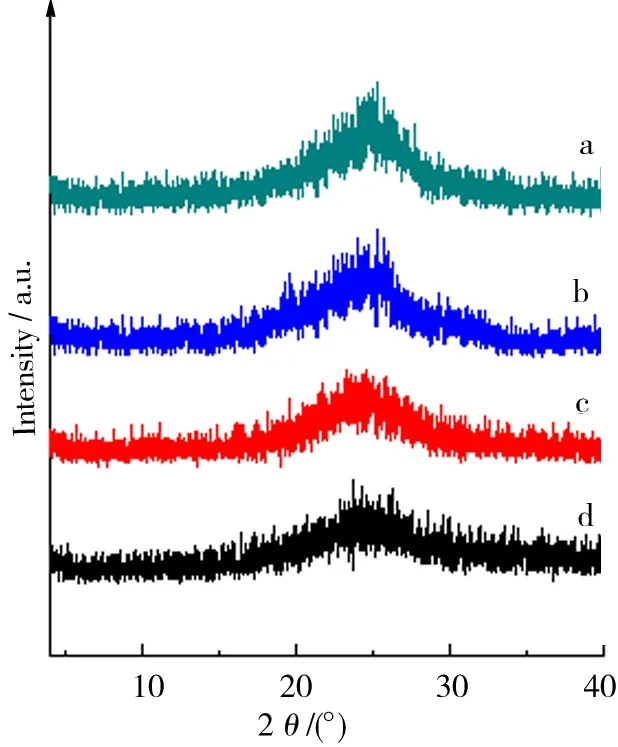
a.GO1;b.GO2;c.GO3;d.GO4.图4 GO还原产物XRD结果Fig.4 XRD results of each GO reduced product
2.4 GO与石墨烯SEM结果
GO与石墨烯SEM结果见图5、图6.由图5可见GO和石墨烯表面的破损、褶皱. 通过颜色可以大致判断GO与石墨烯层数. 碳层堆叠越多,颜色越深. 实验所制得各GO均由多层石墨片层构成. 石墨粒径越小,氧化越充分,剥离程度越高. 各GO的还原产物石墨烯均呈现为褶皱状,粒径越小,剥离程度越高,样品颜色越浅. 图6显示石墨烯的尺寸为10~20 μm,明显高于文献中报道[14-18]. 此结果应源于样品制备中所用石墨粒径大,同时,较长时间的低温预氧化更有利于提高GO粒径.

a.GO1;b.GO2;c.GO3;d.GO4.图5 GO样品的SEMFig.5 SEM results of GO

a.GO1;b.GO2;c.GO3;d.GO4.图6 GO还原后的SEM Fig.6 SEM results of the reduced products of GO
2.5 石墨烯的AFM分析
为了观察GO还原产物石墨烯的单片层厚度,采用AFM对最具代表性的GO1与GO4的还原产物进行了分析. 由图7可见,无论是GO4,还是GO1,其还原产物的厚度均约为1.0 nm. 该厚度是片层上键合的官能团厚度与石墨烯单片层厚度之和. AFM结果证实,GO1与GO4的还原反应完成后都已呈现石墨烯单片层[22].
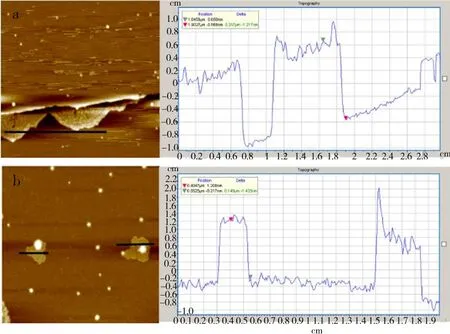
图7 GO1 (a), GO4 (b)的还原产物的AFM结果Fig.7 AFM results of reduced products of GO1(a) and GO4(b)
2.6 GO与石墨烯的Raman结果
物质的Raman光谱特征取决于其分子结构.根据样品的拉曼光谱可得到分子的转动能级与振动能级的信息. 当用一定波长的激光去激发石墨材料时,C环中的sp2杂化原子将产生位于1 650 cm-1的G峰,以及由于晶体缺陷与无序而诱导产生的位于1 360 cm-1处的D峰. 根据G 峰的位置、形状和宽度可确定C的层数与质量.C材料的无序性可以依据D峰与G峰强度之比判断. 从图8、图9所示的GO与石墨烯的Raman光谱可以看出:GO与石墨烯均出现了1 360 cm-1处的D峰与位于1 650 cm-1的G峰. GO1、GO2、GO3、GO4的G峰强度均高于D峰.但是对于其各自的还原产物石墨烯,D峰强度均大于G峰. 这表明GO还原为石墨烯后,其内部结构的紊乱程度增加. 同时,图中结果显示GO与石墨烯的D、G峰强度随石墨粒径的减小而变弱,而其无序性及缺陷增加,转移的质量变多,堆叠层数减少,剥离亦变得越来越充分.
2.7 GO与石墨烯的FTIR结果
图10为GO1、GO2、GO3、GO4的FTIR. 4种GO均产生了对应于-OH、-COOH以及吸附水中-OH的吸收峰(3 700~3 000 cm-1);烷基吸收峰(3 000~2 800 cm-1);C=O吸收峰(1 780~1 680 cm-1);未被氧化的C环吸收峰(1 675~1 520 cm-1);C-OH吸收峰(1 380 cm-1);酯、醚和环氧的C-O吸收峰(1 300~1 000 cm-1). 石墨C粒径越小,其特征吸收峰强度越高. FTIR结果再次证实石墨C粒径越小,其氧化程度越高.
图11为GO1、GO2、GO3、GO4还原产物的FTIR. 由图11结果可以看出,经充分还原后,原GO中的-OH、-COOH的吸收峰(3 700~3 000 cm-1)基本消失,其仅保留了未被氧化的C环的吸收峰(1 675~1 520 cm-1)以及C-O官能团的吸收峰(1 250~1 000 cm-1). 但是,1 250~1 000 cm-1处吸收峰很弱,说明GO还原、脱氧的效果良好[23].
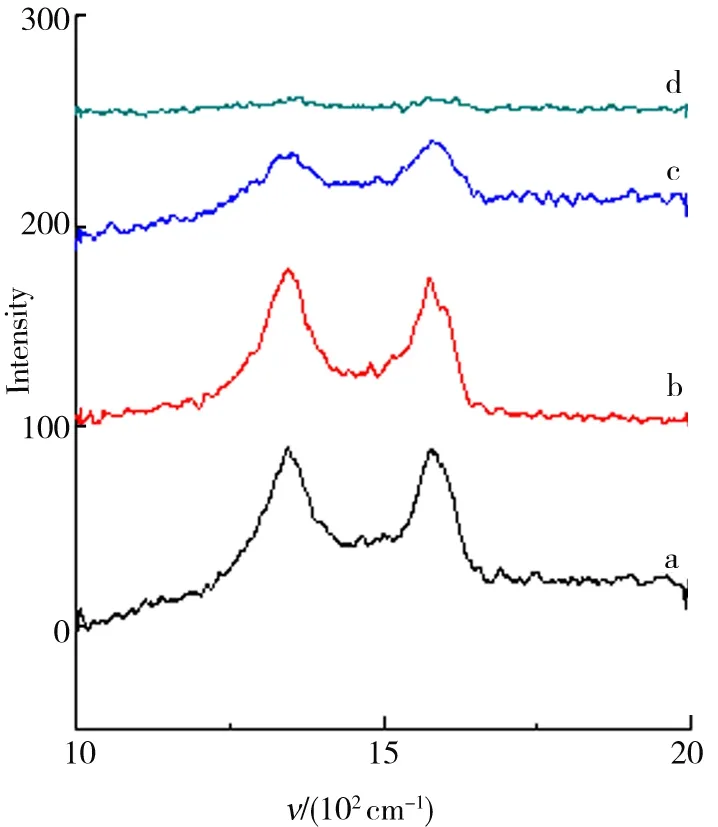
a.GO1;b.GO2;c.GO3;d.GO4.图8 GO Raman光谱结果Fig.8 Raman spectrum results of the GO
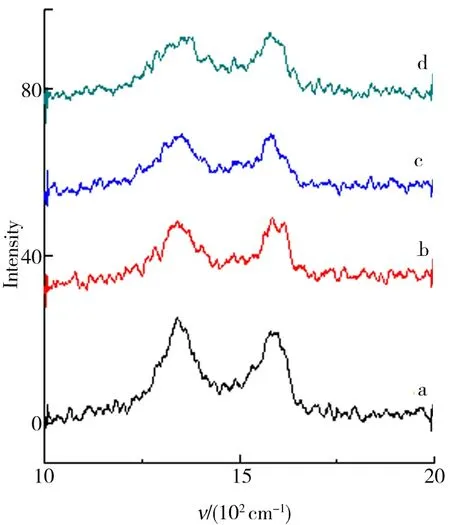
a.GO1;b.GO2;c.GO3;d.GO4.图9 GO还原产物Raman光谱结果Fig.9 Raman spectrum results of the GO reducedproducts
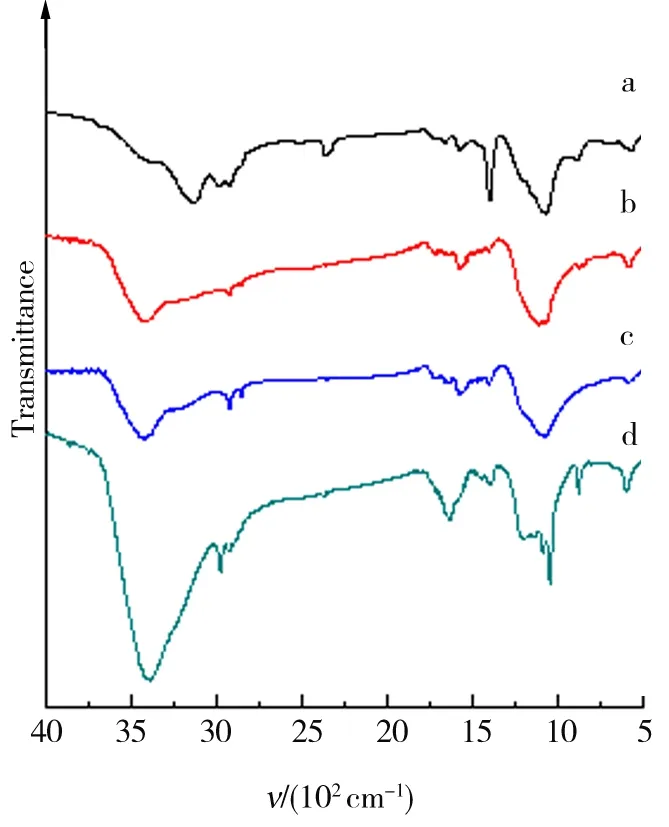
a.GO1;b.GO2;c.GO3;d.GO4.图10 GO的FTIR结果Fig.10 FTIR results of the GO
a.GO1;b.GO2;c.GO3;d.GO4.图11 GO的还原产物FTIR结果Fig.11 FTIR results of reduced products of each GO
2.8 GO与石墨烯的热稳定性分析
采用TG手段考察了GO与石墨烯的质量在升温过程中的变化情况. 由图12可见:由于表面氧化而引入的-OH、-COOH等基团的存在,相比于原料石墨,GO的热稳定性明显下降,并且由于原料石墨粒径越大,氧化反应越困难,因而表现出GO4、GO3、GO2、GO1热稳定性依次增加的趋势.GO显著失重对应温度主要介于200~400 ℃.期间,GO表面上的-OH、-COOH发生热分解释放出H2O、CO和CO2. 400~700 ℃内GO相对稳定,失重缓慢,这是由于更稳定官能团难以去除.图13显示,石墨烯的热稳定性高于GO,其在800 ℃下残炭率均高于80%. 由于还原充分,各石墨烯的热稳定性未表现出明显差异,残炭率均维持在80%~85%.
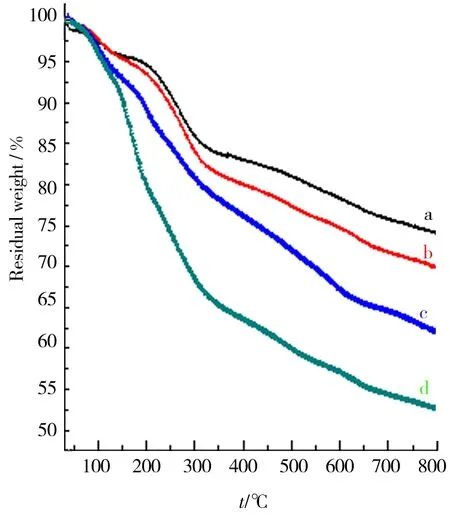
a.GO1;b.GO2;c.GO3;d.GO4.图12 GO的TG结果Fig.12 TG results of GO
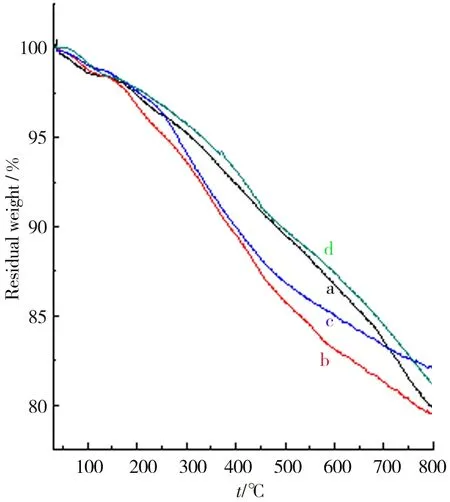
a.GO1;b.GO2;c.GO3;d.GO4.图13 GO还原产物TG结果 Fig.13 TG results of GO reducing products
3 结论
以提高产率,制备大粒径GO和石墨烯为目的,采用密闭氧化、氨-水合胼还原法,以0.10、0.15、0.18、0.30 mm 4种不同粒径石墨为原料分别制备了GO1、 GO2、 GO3、 GO4及其还原产物石墨烯,本研究得到结论如下:
1)分别以0.10、0.15、0.18、0.30 mm石墨为原料,制备各GO的条件为以2.0 g石墨为基准,加入70 mL 浓H2SO4,8.0 g KMnO4,分别在冰浴条件下预氧化8.0 h,80 ℃下加热反应2.0 h,得到的GO产率高,纯度好.
2)冷反应时间是制备高纯度、高产率GO1、GO2、GO3、GO4的关键因素,延长冷反应时间有利于提高GO的产率.
3)以0.10、0.15、0.18、0.30 mm石墨为原料,利用上述方法均可制备尺寸为10~20 μm的石墨烯.
4)石墨粒径越小,所得GO剥离程度、产率越高,但是其热稳定性越差.石墨烯的热稳定性优于GO,其片层厚度约为1.0 nm.
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:48
应用化工(2020年9期)2020-09-29 08:55:16
济南大学学报(自然科学版)(2020年3期)2020-05-21 04:19:12
环球时报(2017-07-19)2017-07-19 11:53:12
中国塑料(2016年7期)2016-04-16 05:25:52
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:36
合成材料老化与应用(2015年4期)2015-07-25 10:45:44
应用化工(2014年1期)2014-08-16 13:34:08
中南民族大学学报(自然科学版)(2014年4期)2014-08-06 05:49:24
城市道桥与防洪(2014年3期)2014-01-08 07:10:10
