
伴CHD7 基因突变的男性性腺功能减退1 例报告并文献复习
2020-07-17 05:59刘贵中吴宝军白文俊牛远杰
中国男科学杂志 2020年3期
刘贵中 吴宝军 白文俊 牛远杰
1.天津医科大学第二医院 泌尿外科,天津300211; 2.天津市津南医院 泌尿外科,天津300350;3.北京大学人民医院 泌尿外科,北京,100044
男性低促性腺功能减退症(HH)病变部位在下丘脑或垂体,多由基因突变引起,就诊者首先检查垂体MR 除外垂体结构性病变,担心后代遗传问题行基因测序明确病因。男性HH 患者LH 和FSH 减低,睾酮缺乏,青春期发育不明显或不完全,常常伴有小阴茎、小睾丸、阴毛稀疏、隐睾或尿道下裂等,早期积极临床干预,预后良好。
病历资料
一、病史
患者男性,23 岁,因发育不良于2019年3月11日就诊,有包皮环切史。身高171cm,体重75Kg,嗅觉正常,胡须稀疏;Tanner 分期:阴毛P3 生殖器G3;双侧睾丸体积1~2ml,质地软,阴茎牵拉长度(SPL)7cm。
二、辅助检查
1、左手骨龄片:骨龄17 岁;
2、阴囊超声:双侧睾丸小,左侧睾丸15mm*12mm*8mm,右侧睾丸16mm*10mm*9mm,双侧附睾小,彩色血流信号未见异常。
3、垂体MR:垂体稍小。
4、性激素水平(见表1)。
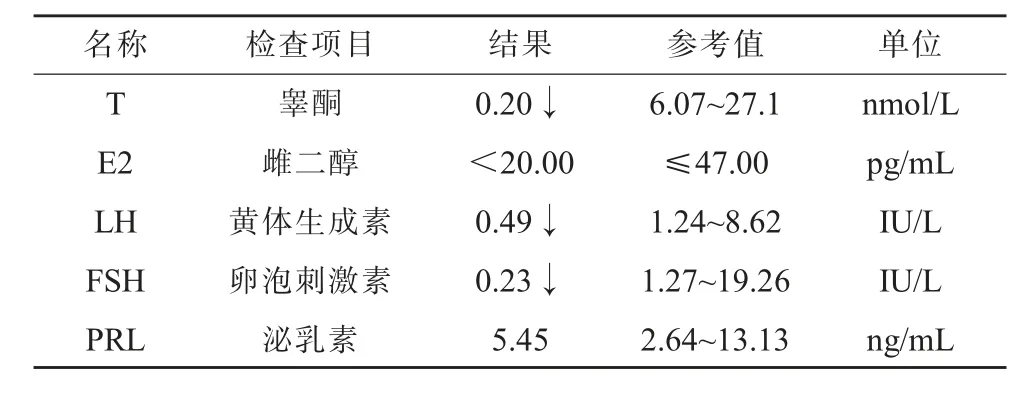
表1 性激素五项水平
5、全基因外显子测序
(1)先证者测序结果

图1 全基因外显子测序
CHD7 基因杂合突变,8 号染色体第33 个外显子编码区6955位的胞嘧啶替换为胸腺嘧啶,导致第2319 位的精氨酸(CCG)转变为半胱氨酸(CTG)
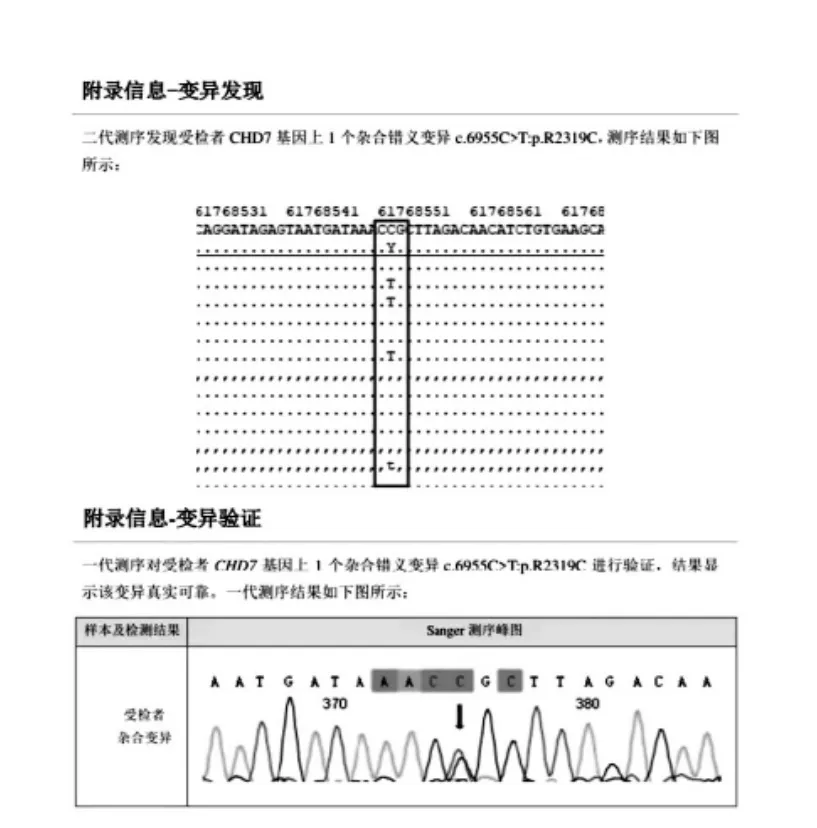
图2 二代测序变异发现和一代测序变异验证
(2)先证者母亲Sanger 测序结果:

图3 先证者母亲Sanger 测序
(3)先证者父亲Sanger 测序结果:

图4 先证者父亲Sanger 测序
三、治疗
注射用绒促性素 (hCG)(丽珠制药) 2000 单位Q72h 肌注;注射用尿促性素(hMG)(丽珠制药)75 单位Q72h 肌注;十一酸睾酮软胶丸(安特尔)80mg Bid,早晚餐中口服。
2020年2月复查骨龄片示骨骺闭合,遂停用十一酸睾酮胶丸口服治疗,继续肌注hCG 联合hMG,鼓励患者尽早结婚生育。
四、随访
上述药物连续应用治疗9 个月后出现精液。随访13 个月,胡须增多,阴毛P5,生殖器G4,SPL 10cm,阴茎晨勃及夜间勃起明显,性欲强,左侧睾丸约11ml,右侧睾丸约12ml。2020年4月精液常规:量1.5ml,PH7.3,精子浓度10.2×106/mL,活力32%。
讨 论
CHD7 基因位于染色体8q12.1,包含38 个外显子,大小约188kb,编码2997 个氨基酸,CHD7 包含多个结构域,属于ATP 依赖的染色质解旋酶DNA 结合蛋白家族,与其他分子协同发挥染色质重塑作用[1]。CHD7是染色质结构域解螺旋酶DNA 结合蛋白7 基因,具有水解ATP 的能力,改变核小体结构,CHD7 基因编码的蛋白质通过修饰染色质促进转录调控和胚胎发育,在染色质重塑、转录调控、细胞周期调控、细胞凋亡和胚胎干细胞调控等方面具多种功能[2]。致病性CHD7 等位基因错义突变破坏ATP 酶和核小体重塑活性,可能是导致HH 发生的分子机制[3]。
CHD7 在胚胎组织细胞广泛表达,不仅影响神经系统和生殖系统发育,而且对下丘脑、嗅球、耳、心脏、骨骼等器官的生长发育有重要的意义,胚胎时期CHD7在下丘脑、嗅球嗅束、垂体前叶、中叶细胞呈高表达,该基因突变导致GnRH 神经元不能正确的迁移及嗅球发育不良,发生Kallmann 综合征(KS)[4];CHD7 基因突变致下丘脑GnRH 神经元分泌和作用缺陷,发生低促性性腺功能减退症(HH)[5,6]。Kim[5]对101 例HH/KS 患者进行CHD7 的38 个外显子进行筛选,发现3 例散发性KS 和4 例散发性IHH 存在不同程度的CHD7 杂合突变,2 例为点突变,5 例为错义突变,影响高度保守的染色体结构域,CHD7 基因突变导致单倍剂量不足是发生HH/KS 的主要分子机制。
CHD7 突变引起CHARGE 综合征[7],为常染色体显性遗传疾病,伴有听力受损、耳畸形、唇腭裂、心脏缺陷、后鼻道闭锁、生殖器发育不良(如隐睾、小阴茎、尿道下裂、子宫缺如等)和生长发育迟缓等[8,9]。Siavriene[10]报道CHD7 基因突变导致氨基酸移码突变发生CHARGE 综合征;Wu[8]对177 例HH 患者进行测序,发现10.2%携带CHD7 基因突变。HH 是由于促性腺激素分泌不足导致青春期不发育或发育不全,表现为睾丸发育不良、小阴茎、生精功能障碍等[11],与CHARGE 综合征有重叠的临床表型特征,但不伴有心脏、耳、鼻等异常表型,仅仅是影响了GnRH 神经元向下丘脑的迁移或合成与释放[12]。
CHD7 突变来源分为家族遗传性和新发突变两种,家族遗传性是父母亲双方或一方携带相同的致病突变基因,而新发突变是父母亲均不携带相同的致病突变基因。新发基因突变可能发生在胚胎受精卵卵裂时期,与一些物理因素(射线、紫外线等)、化学因素(亚硝酸盐、金属离子、生物碱、抗生素、农药等)或生物因素(细菌、病毒感染)等影响有关。本例HH 患者因发育不良就诊,发现小阴茎、小睾丸、胡须和阴毛稀疏,通过全基因外显子测序发现CHD7 杂合突变,未发现其他可疑致病基因突变,考虑CHD7 杂合突变为HH 致病原因。对先证者父母进行Sanger 测序未见CHD7 杂合突变,可除外家族遗传性,遂考虑为新发致病基因突变。CHD7 基因发生错义突变,精氨酸转变为半胱氨酸,致使蛋白结构域或功能发生改变,影响大脑和神经系统发育,GnRH 神经元迁移障碍或合成作用缺陷,促性腺激素分泌不足,睾酮降低,导致男性第二性征发育不良。患者就诊时骨龄17 岁,几乎接近闭合,骨骺闭合后阴茎将停止继续生长发育[13],因而治疗时间迫切,遂应用双促联合外源性睾酮补充治疗,双管齐下(促性腺激素+睾酮),尽快改变患者一穷(小阴茎)二白(胡须、阴毛稀疏)的面貌,促进睾丸发育及精子成熟,促进男性第二性征发育。
男性HH 的治疗以补充睾酮为主,骨骺闭合前早期用药促进男性第二性征发育,改善男性第二性征不足[15]。有生育需求者,建议应用hCG 联合hMG 治疗,促进精子发生,提高自然妊娠率[16];外源性补充睾酮维持男性身体机能及性功能,适合于无生育需求HH 患者。CHD7 基因突变报道为常染色体显性遗传[14],单基因病遗传概率符合孟德尔定律,自然妊娠后代约50%概率发生HH,如担心后代遗传问题,建议选择胚胎植入前遗传学筛查,做到优生优育。
猜你喜欢
现代泌尿生殖肿瘤杂志(2022年1期)2022-11-21
临床输血与检验(2022年3期)2022-06-22
种子(2021年3期)2021-04-12
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中国体育教练员(2017年3期)2018-01-19
中国体育教练员(2017年2期)2017-07-31
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
大众健康(2016年3期)2016-05-31
