
Mn掺杂Co氧化物用于碱性氧还原与氧析出反应双功能催化剂
2020-07-15 03:24李国强张紫琼麦婉珊张凯凯聂仁峰胡玮
湖北大学学报(自然科学版) 2020年4期
李国强,张紫琼,麦婉珊,张凯凯,聂仁峰,胡玮
(有机化工新材料湖北省协同创新中心,有机功能分子合成与应用教育部重点实验室,湖北大学化学化工学院,湖北 武汉 430062)
0 引言
严峻的能源与环境问题促使人们开发清洁且可再生能源,多种电化学能源存储与转换装置,如可充放电金属-空气电池,一体化可再生燃料电池等[1-4]得到了广泛的关注,但其中的关键步骤——氧电催化过程,包括氧还原反应(ORR)及氧析出反应(OER),却制约了其能量转化效率.ORR与OER涉及4电子反应过程,本质复杂且具有慢动力学特征,因而需要高的超电势来提升其反应效率[5].迄今为止,Pt基催化剂依然是ORR的最佳催化剂,而OER则依赖于贵金属Ir或Ru基材料,丰度低、成本高使其商业化应用受到很大限制[6-8].此外,Pt和Ir均不能同步催化OER和ORR,而可再生能源装置却迫切需要双功能催化剂.不过,一些非贵金属替代材料,如尖晶石型或钙钛矿型混合价态过渡金属氧化物等[9-10]有望打破这种约束[11-13].
近年来,通过以下三种策略,廉价的ORR/OER双功能催化材料取得了很大的研究进展.一是通过阳离子掺杂来调节主体阳离子的电子结构,改变其原来不利于ORR/OER的过低或过高的价态[14-15].如Co3O4在碱性介质中对OER具有良好的催化活性和稳定性[16],但导电性略差,通过掺入其他金属形成AB2O4化合物如NiCo2O4与MnCo2O4等,不仅提升了导电性与稳定性,而且加速了含氧中间体的吸/脱附速率,从而提升了OER性能.在掺杂的同时进行形貌和结构可控的合成方法学研究是第二种策略.由于CoxMn3-xO4的晶相对金属配比十分敏感,因此通过合成方法同步调节晶相和组成是一个巨大的挑战.从晶体结构上看,AB2O4尖晶石结构为氧离子按立方紧密堆积排列,A2+离子充填于1/8的四面体空隙中,B3+离子充填于1/2的八面体空隙中,因而具有独特的电活性B3+/B2+与A3+/A2+氧化还原电对[17-18].陈等[19]用“氧化沉淀-嵌入晶化”两步法实现了晶相与组成的同步调节,并在常压和较低温度下成功合成了系列MxMn3-xO4(M为Co,Mg及Zn等二价金属)尖晶石超细纳米材料.他们发现立方相Co-Mn-O尖晶石结构的ORR本征活性高于四方相,而后者的OER性能则高于前者,并证明了Co-Mn氧化物对ORR的本征活性与晶相、Co/Mn比和表面价态有关,这些因素会影响O2的活化程度和暴露的活性位点数量.第三类策略是利用碳基材料,尤其是杂原子掺杂碳与CoxMn3-xO4的显著协同效应.CoxMn3-xO4与碳基材料的化合耦合作用可以有效提升催化性能,如Zhao等[20]报道了一种Mn-Co氧化物/氮掺杂多壁碳纳米管杂化材料(MnxCo3-xO4@NCNTs),其ORR活性可以媲美商业Pt/C,OER与ORR的电势差值仅有0.93 V.碳基材料的载体效应,提供了高分散率,防止催化剂在电化学反应中聚结或损失;CoxMn3-xO4和杂原子掺杂C优异的本征活性;N或其他含杂原子掺杂碳与CoxMn3-xO4间的强烈的化学耦合作用可以给予很好的解释.
可见,找到具有ORR/OER双功能活性的Co-Mn氧化物的有效制备途径,了解它们的性能与多价态、合适的晶体结构和独特形貌间的关系仍然非常必要.本工作基于水热法成功制得Mn掺杂的Co-Mn氧化物纳米晶体,然后结合晶相、结构、化学成分或价态对其双功能催化活性进行了考察,并解释了Mn的加入对所得材料催化性能的影响机制.
1 实验部分
1.1 Co-Mn氧化物催化剂的制备乙酸钴与乙酸锰共6 mmol溶解于20 mL去离子水中,缓慢加入10 mL NaOH溶液(1.2 mol/L),搅拌3 h后转移至50 mL反应釜,水热反应180 ℃,12 h后,经水洗、干燥后得灰褐色固体.随后在Ar气氛下,以5 ℃/min升温至500 ℃热处理3 h,得到黑色CoxMn1-xO粉末.调节Co/Mn摩尔比分为1∶0、1∶1、2∶1及3∶1,分别制得CoO,Co0.5Mn0.5O,Co0.66Mn0.33O与Co0.75Mn0.25O样品.
1.2 实验仪器X线衍射仪(XRD,Bruker D8Advance,40 kV加速电压);场发射扫描电子显微镜(FESEM,JEOL,JSM-6700F,5 kV);透射电子显微镜(TEM,JEOL JEM-2010);X线光电子能谱仪(XPS,Kratos Ltd.XSAM800);电化学工作站(上海辰华,CHI660E);旋转圆盘电极装置(美国Pine).
1.3 电化学性能测试将催化剂分散于含Nafion的异丙醇溶液中超声分散形成墨水(5 mg/mL),涂覆在预先抛光的玻碳电极(GC)上室温干燥形成均匀的催化剂薄膜,并以此为工作电极,以铂片电极为对电极,Hg/HgO为参比电极,在0.1 mol/L KOH溶液中进行测试.
2 结果与讨论
2.1 Co-Mn氧化物的物理表征XRD 表征样品的晶体结构发现(图1a),除Co/Mn=1∶1氧化物外,Mn含量较低或不含Mn的Co氧化物(如1∶0,2∶1与3∶1)样品具有相似的特征,其衍射峰分别与立方相CoO(PDF#43-1004)的(111)、(200)、(220)、(311)及(222)晶面对应,未发现分相的Mn氧化物,说明均形成单一立方晶相[21].由相应的局部放大谱图(图1b)可知,Co/Mn比例为3∶1与2∶1氧化物的两个主要特征峰位置相对于单金属CoO依次略有左移,这是由于Mn掺杂进入了CoO晶格,且Mn离子半径(如Mn2+,r=0.080 nm)大于Co离子半径(如Co2+,r=0.072 nm),造成了晶格膨胀.根据(200)晶面由谢乐公式估测晶粒尺寸依次为:CoO(35 nm),Co0.75Mn0.25O(19 nm)与Co0.66Mn0.33O(14 nm),说明随着Mn含量的增加,晶粒尺寸明显下降.不过,氧化物中Mn含量过高,如Co/Mn为1∶1时,除了CoO晶相外,有明显四方相[(Co,Mn)(Mn,Co)]2O4(PDF#18-0408)结构存在,说明过量Mn加入打破了原有的CoO晶体结构,形成了分相的氧化物.
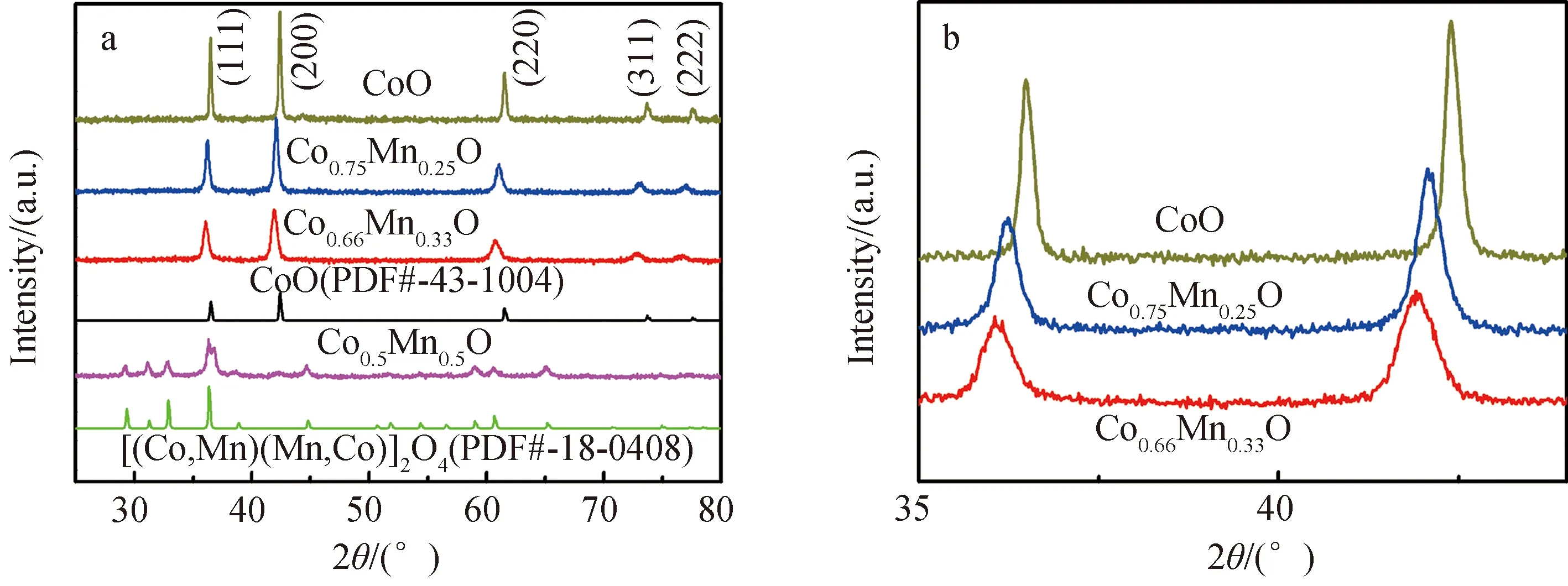
图1 (a)Co-Mn氧化物的XRD谱图;(b)局部放大XRD谱图
进一步用 XPS分析CoO,Co0.66Mn0.33O与Co0.75Mn0.25O样品的表面化学状态及组成,如图2所示.3种样品的Co2p谱图(图2a)均由Co2+及Co3+构成,并伴随着两对振激卫星峰;Mn2p谱图(图2b)分别对应Mn2+、Mn3+及Mn4+氧化态;O1s谱图(图2c)对应晶格氧(如Co-O)、化学吸附氧(如—OH)及物理吸附H2O,相应的结合能位置见表1[1].双金属氧化物(Co0.66Mn0.33O与Co0.75Mn0.25O)的Co2p峰位置相对于单金属CoO分别右移了0.6~0.3 eV,说明Mn的加入改变了Co的电子结构,使Co核外电子向Mn偏移.由于过渡金属离子的电子结构对其表面含氧物种的吸附能力有显著影响,从而可以影响电催化OER/ORR的性能[22-23].金属氧化物中的多价3d过渡金属由于通过电子跃迁而增强了电子传导,以及氧化还原反应介导的电荷转移的增强,对电催化性能也有促进作用.特别是,在eg轨道上有单个电子的过渡金属(如Mn3+的t3e1和Co3+的t5e1)被认为是氧电催化反应更活跃的位点[24].Co0.66Mn0.33O与Co0.75Mn0.25O中Co离子与Mn离子平均价态分别为2.42,2.93,其中Mn3+/Mn4+是有效的ORR活性位点,Co3+对OER有益,因此在四面体中自由分布的Co、Mn阳离子可以平衡OER与 ORR活性,从而保证了催化剂的双功能活性[25].表2列出了XPS及EDX定量分析结果,其中EDX表征Co0.66Mn0.33O与Co0.75Mn0.25O样品中Co/Mn摩尔比分别为2.1∶1和3.0∶1,与投料比一致;相应的XPS所得表面Co/Mn摩尔比分别为1.5∶1和1.8∶1,略低于对应的体相组成,说明近表面处Mn含量偏高,可能是由于Mn阳离子在热处理成相时的向外扩散速率高于Co阳离子,从而形成了富含Mn的表面结构[26].
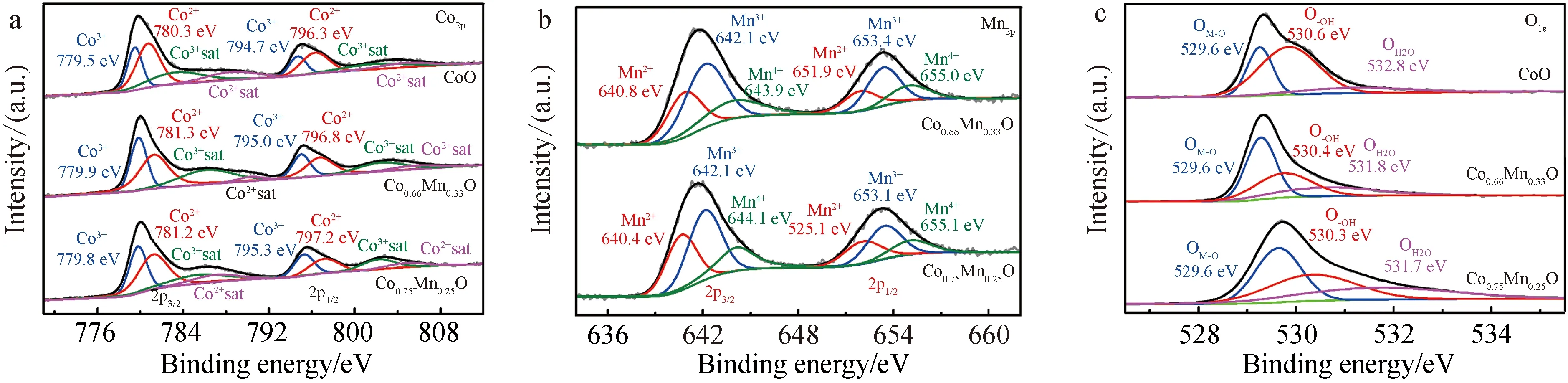
图2 CoO、Co0.66Mn0.33O与Co0.75Mn0.25O样品的Co2p(a),Mn2p(b)及O1s(c)XPS谱图
SEM观察Co0.75Mn0.25O与Co0.66Mn0.33O样品(图3)发现,二者均为不规则微粒的聚集体,微粒之间可见堆积孔,对应的平均尺寸分别为(89.2±16.6)nm及(74.5±16.4)nm.进一步用TEM观察Co-Mn氧化物与CoO的区别(图4),发现CoO为不规则的球状或方形颗粒,尺寸不一(77~125 nm),但含Mn的双金属氧化物的微粒尺寸明显下降,其中Co0.75Mn0.25O也为不规则方形或球状形貌,而Co0.66Mn0.33O则为花球状形貌.

表1 对CoO,Co0.66Mn0.33O与Co0.75Mn0.25O样品的XPS分析结果

表2 CoO,Co0.66Mn0.33O与Co0.75Mn0.25O样品的XPS及EDX元素分析结果

图4 CoO(a)、Co0.75Mn0.25O(b)与Co0.66Mn0.33O(c)样品的TEM图
2.2 Co-Mn氧化物的电化学性能由CV图(图5a)可知,CoO,Co0.75Mn0.25O与Co0.66Mn0.33O催化剂在O2气中的CV曲线均有明显的还原峰,与Ar气下的情况明显不同,说明该还原峰均由O2的电化学还原过程引起;从ORR的极化曲线(图5b)及相应的参数:半波电势(E1/2)及0.7 V下的动力学电流(jk@0.7 V)比较(图5c)看来,Mn元素的掺杂显著提升了CoO本体的催化活性,以Co/Mn摩尔比为3∶1为最佳(E1/2为0.73 V,jk@0.7 V为7.77 mA·cm-2),活性的提升主要得益于Mn对Co的电子调控作用.比较3种催化剂的塔菲尔(Tafel)曲线(图5d),发现Tafel斜率依次为145.7 mV·dec-1(CoO),136.0 mV dec-1(Co0.66Mn0.33O)及100.0 mV·dec-1(Co0.75Mn0.25O),表明Mn加入也加快了反应动力学.由不同转速下的极化曲线及对应的Koutecky-Levich(K-L)图(图5e、图5f及图5g)分析可知,CoO,Co0.75Mn0.25O与Co0.66Mn0.33O的电子转移数(n)分别为3.60,4.01及4.25,表明Mn的加入提高了Co基催化剂对氧还原4电子过程的选择性,Co0.66Mn0.33O的n大于4.0,可能是由于在选取的计算电势范围内(0.40~0.55 V)样品电流密度差别较小引起.

图5 CoO、Co0.66Mn0.33O与Co0.75Mn0.25O的电催化ORR性能比较
上述催化剂也有明显的OER性能(图6a),其中以Co0.75Mn0.25O的OER活性最佳,在10 mA·cm-2的电势为1.699 V,低于Co0.66Mn0.33O及CoO上所得到的结果(均为1.733 V),与文献报道结果接近(表3),说明适量的Mn掺杂(Co/Mn=3条件下)不仅可以作为ORR的活性位点,也能维持Co位点的OER活性,并通过Co-Mn离子间的协同作用,调控整个催化剂的性能.图6b显示,3种材料的Tafel斜率并未产生明显的变化,说明其反应历程基本相似,但Tafel斜率由CoO的138.8 mV·dec-1略降为Co0.75Mn0.25O的131.5 mV·dec-1,说明Mn的加入一定程度上提升了反应速率.经历2 500圈循环电势扫描后(图6c),3种催化剂的电流密度均有下降,不过Co0.75Mn0.25O催化剂上的电势(10 mA·cm-2)仅右移了约18 mV,明显优于CoO的稳定性.可见,Mn掺杂同时也增强了Co基催化剂的稳定性.
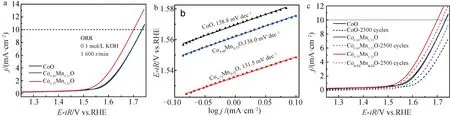
图6 CoO、Co0.66Mn0.33O与Co0.75Mn0.25O的电催化OER性能比较
此外,表3还比较了Co0.75Mn0.25O与文献报道典型Co或Mn基催化剂的双功能活性,用OER与ORR电势差,即ΔE(ΔE=EOER(j=10)-EORR(j=-3))来衡量。其中,Co0.75Mn0.25O的ΔE为1.00 V,优于CoMn2O4(1.08 V)[27]、Co3O4/Co2MnO4(1.09 V)[28]、Co-Mn氧化物/N掺杂C纳米纤维复合物(1.06 V)[29]及贵金属20% Pt/C(1.17 V)[30],但比Co-Mn-Ni三元氧化物纳米棒(CMN-231,0.88 V)[31]及Co3O4/N-HNMK-3(0.86 V)[32]略差.可见,Mn掺杂CoO可作为有竞争力的非贵金属双功能催化剂,有望在能源转化与存贮装置中展开应用,基于上述研究结果的交联多孔纳米新结构Co-Mn氧化物的合成是下一步研究的重点。

表3 CoO、Co0.66Mn0.33O与Co0.75Mn0.25O的电催化性能比较 V
3 结论
本研究采用简单水热法制备了Mn掺杂的CoO纳米晶体,发现Co0.75Mn0.25O与Co0.66Mn0.33O材料具有较好的双功能催化活性,且Co0.75Mn0.25O更为突出,其催化ORR的E1/2为0.73 V,使OER达到10 mA·cm-2仅需要提供469 mV超电势,明显优于仅含单一金属的CoO催化剂,且稳定性也有显著提升.Mn的加入不仅增加了有效的反应活性位点,并且合适的Co/Mn原子摩尔比及Mn-Co间的电子相互作用,能明显调控催化剂中过渡金属阳离子核外电子分布,从而改善其表面对反应含氧物种的吸附能力,提升反应活性.
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
学校教育研究(2021年20期)2021-12-14
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
防爆电机(2020年4期)2020-12-14
分析化学(2018年12期)2018-01-22
分析化学(2017年1期)2017-02-06
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
山东工业技术(2016年15期)2016-12-01
中学化学(2015年9期)2016-04-14
