
SCN1A基因突变类型与Dravet综合征患儿临床特征的相关性
2020-07-14 10:57陈易得陈金妮
山西医科大学学报 2020年6期
陈易得,张 华,陈金妮
(三亚中心医院,海南省第三人民医院儿科,三亚 572000)
Dravet综合征(Dravet syndrome,DS)作为少见的婴幼儿发育性癫痫性脑病,主要于1岁内发病,尤其是生后6个月内为发病高峰年龄,发热引起的较长时间惊厥发作是其首发症状,临床表现为双侧强直阵挛发作、单侧肢体阵挛,临床控制难度大[1,2]。研究指出,随着临床分子遗传学技术不断发展、临床深入分析DS发现,遗传因素于DS发病机制中发挥重要作用,其中SCN1A编码电压门控钠离子通道(VGSC)相关α亚单位发生突变为DS发病主要原因,可造成大脑神经元细胞功能障碍,干扰脑功能正常发育,诱发癫痫[3,4]。研究数据显示,众多DS患儿发病主要是SCN1A基因突变引起,且多数是新生突变,突变位点、突变类型呈多样化,其中突变类型主要表现为错义突变、截断突变(剪切突变、无义突变、大片段缺失/重复、移码突变),而错义突变多集中在门孔区(S5-S6)、电压感受区(S4)[5]。相关研究指出,临床对SCN1A突变类型的识别已超过1 200种,其中>900种突变类型和DS发病有关,但针对SCN1A突变类型和DS临床特征是否有关仍缺乏报道[6]。基于此,本研究对SCN1A基因突变类型与Dravet综合征(DS)患儿临床特征的相关性进行分析,以指导临床早期识别和诊治。
1 资料与方法
1.1 一般资料
选择2016-09~2019-08本院儿科收治的47例DS患儿作为观察对象,其中男26例,女21例,年龄是1-8岁,平均年龄是(4.67±1.09)岁。纳入标准:入选患儿全部通过临床检查判定是Dravet综合征,符合DS诊断标准;临床资料登记完整;患儿家长全部知情,签署同意书;获本院医学伦理委员会审批。排除标准:临床资料、随访信息未完善患儿;癫痫发作前产生精神运动发育迟缓者;伴遗传代谢病、肿瘤及外伤等病因明确的癫痫;发作类型中出现失张力发作、强直发作患儿;围产期发生严重性低血糖、颅内感染、缺氧缺血、脑梗死、先天性脑发育畸形等相关神经系统损害患儿;发作和热敏感性相关性未确定及无显著性患儿;未同意接受基因检测相关患儿。
1.2 方法
1.2.1 采集病史资料 帮助患儿准确创建临床资料登记表,涉及患儿性别、出生日期、发育状况、性别、围生期状况、家族史、初次发病年龄、重要辅助检查、疾病发作类型/频率、用药既往史、用药治疗效果、不典型失神与肌阵挛的发作产生时间等情况,经面谈或是电话询问患儿家长,对患儿病史资料进行详细、完整登记。
1.2.2 基因检测 抽取患儿、家系成员2 ml外周血,放入抗凝试管(内含乙二胺四乙酸)中,并送至实验室开展基因检测。首先通过二代高通量目标区域准确捕获测序实施癫痫基因测定;再经人类基因突变数据库和千人基因组数据库等相关数据库分析数据,借助蛋白损伤相关预测软件预测突变,探索和疾病有关的可疑致病性、致病性基因突变。通过Sanger测序法验证分析患儿家长家系,针对缺乏明确致病性基因突变患儿,通过MLPA技术检测SCN1A基因的大片段变异。
1.2.3 智力发育评估 对入选患儿开展智力发育评估,由院内专业的、资深的智力测评医师开展评估,通过Gesell量表对≤6岁患儿实施智力评估,通过C-WISC智能量表对>6岁患儿的智力实施评估,并参照评估分数分成正常智力及轻度、中度、重度、极重度智力低下[7,8]。
1.3 统计学分析
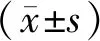
2 结果
2.1 临床发作特点
47例DS患儿的初次发病年龄是3-14个月,平均发病年龄是(6.00±1.35)个月;32例患儿首次发作是发热诱发;15例患儿首次发病是无热惊厥,后伴热敏感性;28例患儿于病程中伴热水浴惊厥发作情况;35例患儿存在惊厥持续状态,持续时间是0.5-3.4 h;38例患儿出现丛集样发作,即24 h内抽搐次数≥2次;43例患儿发作类型≥2种,44例患儿呈强直阵挛发作,26例患儿呈不典型失神发作,21例呈全面性阵挛发作,33例患儿呈肌阵挛发作,5例患儿呈强直发作,35例呈局灶性发作。
2.2 智力发育评估结果
47例患儿中智力发育正常、轻度智力低下、中度智力低下、重度智力低下检出率分别是17.02%(8/47),29.79%(14/47),38.30%(18/47),14.89%(7/47)。
2.3 基因测定结果
47例患儿中SCN1A基因突变阳性36例,突变发生率是76.59%,包括18例(50.00%)错义突变、18例(50.00%)截断突变,截断突变包括3例(8.33%)剪切突变、10例(27.78%)移码突变、2例(5.56%)大片段缺失及3例(8.33%)无义突变。错义突变中13例患者处于S4-S6区(编码核心区)、5例患儿处于其他编码区(非S4-S6区),且5例患儿中存在相同基因突变位点者3例(见表1、图1)。35例患儿是新生突变,1例患儿突变源自母亲。27例患儿的SCN1A基因突变是既往未提出新发突变,9例患儿的突变存在既往致病性报道,为错义突变(见图2)。
2.4 SCN1A基因突变类型和临床特征关系
错义突变组DS患儿的不典型失神发作年龄、肌阵挛发作年龄均高于截断突变组DS患儿,但丛集样发作率低于截断突变组,差异有统计学意义(P<0.05);错义突变组、截断突变组DS患儿的性别、首次发病年龄、惊厥持续状态发生率、癫痫发作类型、智力发育评估结果对比,差异无统计学意义(P>0.05,见表2)。
表1 47例DS患儿基因测定结果
Table 1 Gene test results of 47 children with DS

突变类型 例数(%) 错义突变 核心编码区13(36.11) 其他编码区 5(13.89)截断突变 剪切突变 3(8.33) 移码突变10(27.78) 大片段缺失 2(5.56) 无义突变 3(8.33)SCN1A基因突变36(76.59)
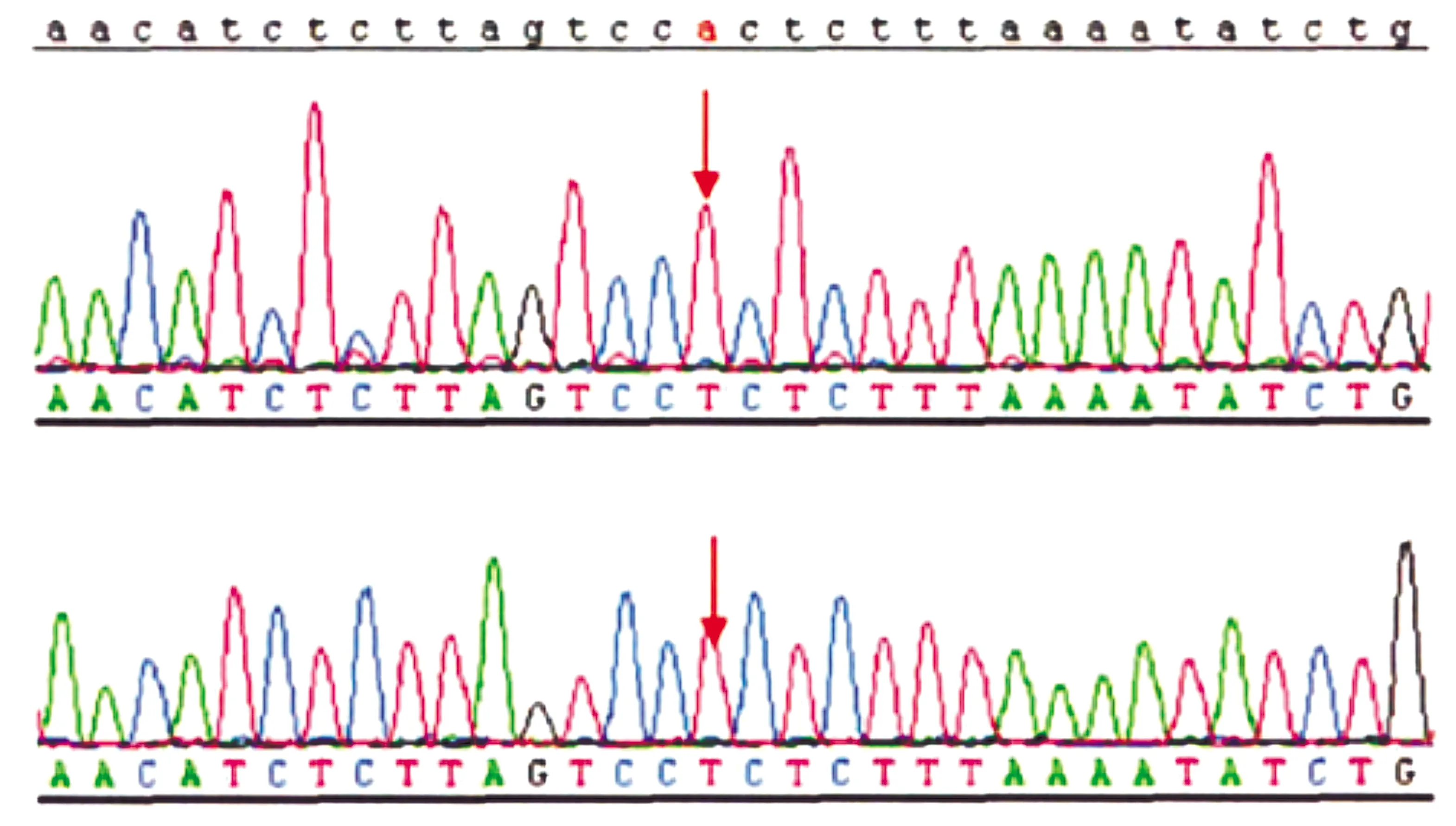
核苷酸变化为c.2836C>T,患儿中存在相同基因突变位点图1 DS患儿SCN1A基因测序图Figure 1 SCN1A gene sequencing diagram of children with DS

氨基酸变化为p.(Arg946Cys),为错义突变图2 DS患儿SCN1A基因杂合变异Figure 2 SCN1A gene heterozygous variation of a DS child
表2 错义突变、截断突变DS患儿临床特征
Table 2 Clinical characteristics of children with missense mutations and truncated mutations in DS

临床特征错义突变(n=18)截断突变(n=18)χ2/tP性别0.1140.735 男 10(55.56) 11(61.11) 女 8(44.44) 7(38.89)首次发病年龄(月)6.38±3.54 5.20±1.37 1.3190.196不典型失神发作年龄(月)25.96±9.15 16.79±5.83 3.5860.001肌阵挛发作年龄(月)21.14±9.17 14.76±5.92 2.4800.018丛集样发作 11(61.11) 17(94.44)5.7860.016惊厥持续状态 15(83.33) 13(72.22)0.1610.688发作类型 强直发作 1(5.56) 2(11.11)0.0001.000 肌阵挛发作 14(77.78) 11(61.11)1.1780.278 局灶性发作 10(55.56) 15(83.33)3.2730.070 全面性阵挛发作 7(38.89) 8(44.44)0.1140.735 不典型失神发作 11(61.11) 8(44.44)1.0030.317 全面强直阵挛发作 17(94.44) 17(94.44)0.0001.000智力发育评估 正常 4(22.22) 2(11.11)0.2000.655 轻度低下 5(27.78) 5(27.78)0.0001.000 中度低下 6(33.33) 7(38.89)0.1200.729 重度低下 1(5.56) 2(11.11)0.0001.000
2.5 不同编码区SCN1A基因错义突变患儿临床特征
S4-S6区、非S4-S6区SCN1A基因错义突变患儿的首次发病年龄、不典型失神发作年龄、肌阵挛发作年龄、丛集样发作发生率、惊厥持续状态发生率、癫痫发作类型、智力评估结果对比,差异无统计学意义(P>0.05,见表3)。
表3 S4-S6区、非S4-S6区SCN1A基因错义突变患儿临床特征
Table 3 Clinical characteristics of children with missense mutations ofSCN1Agene in S4-S6 and non-S4-S6 regions
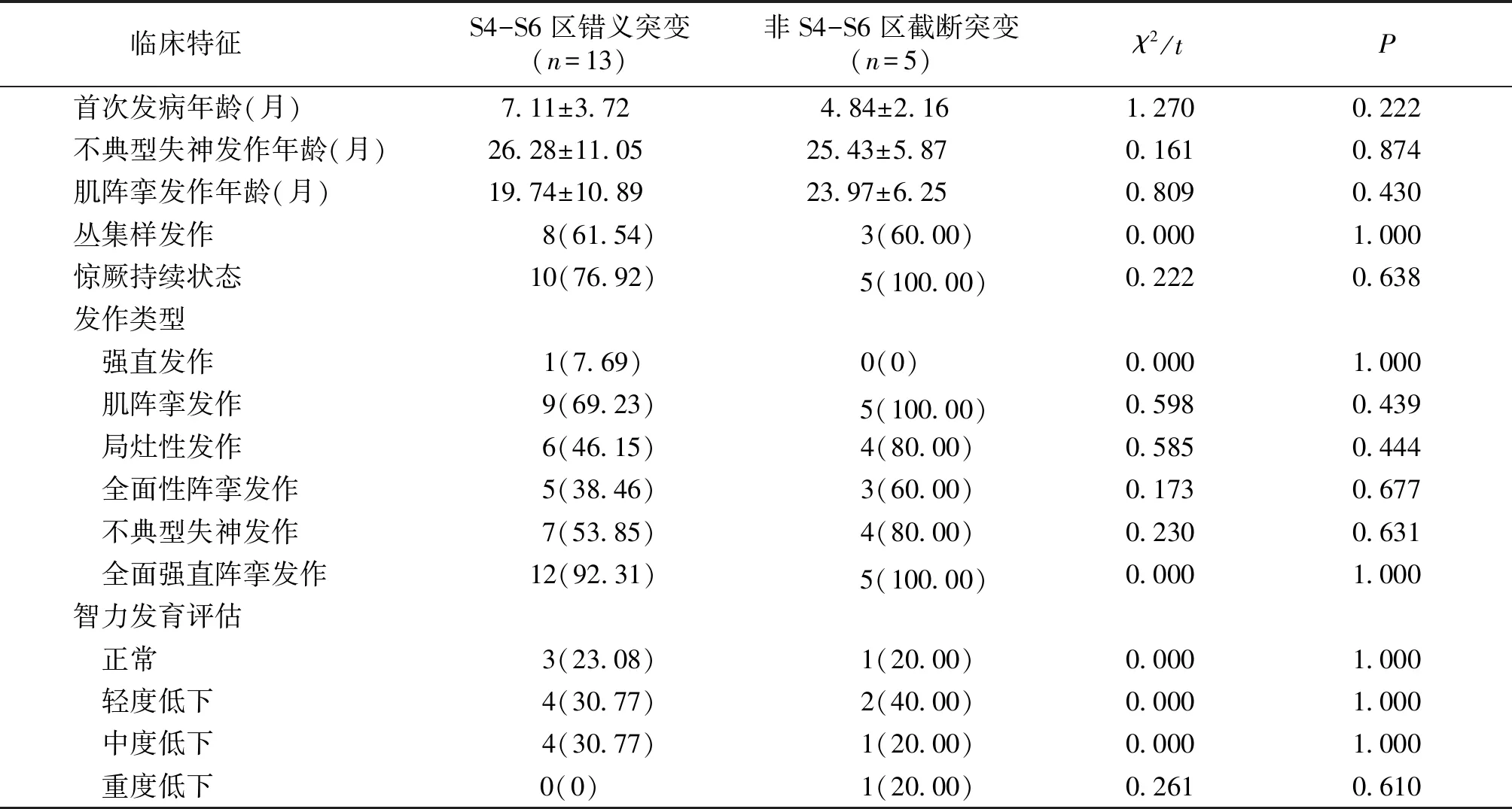
临床特征S4-S6区错义突变(n=13)非S4-S6区截断突变(n=5)χ2/tP首次发病年龄(月)7.11±3.72 4.84±2.16 1.2700.222不典型失神发作年龄(月)26.28±11.05 25.43±5.87 0.1610.874肌阵挛发作年龄(月)19.74±10.89 23.97±6.25 0.8090.430丛集样发作 8(61.54) 3(60.00)0.0001.000惊厥持续状态 10(76.92) 5(100.00)0.2220.638发作类型 强直发作 1(7.69)0(0)0.0001.000 肌阵挛发作 9(69.23) 5(100.00)0.5980.439 局灶性发作 6(46.15) 4(80.00)0.5850.444 全面性阵挛发作 5(38.46) 3(60.00)0.1730.677 不典型失神发作 7(53.85) 4(80.00)0.2300.631 全面强直阵挛发作 12(92.31) 5(100.00)0.0001.000智力发育评估 正常 3(23.08) 1(20.00)0.0001.000 轻度低下 4(30.77) 2(40.00)0.0001.000 中度低下 4(30.77) 1(20.00)0.0001.000 重度低下0(0) 1(20.00)0.2610.610
3 讨论
Dravet综合征又称婴儿严重肌阵挛性癫痫综合征,致病机制复杂,会诱发患儿脑部神经元异常放电,临床主要表现为短暂、重复性惊厥症状,合并脑部中枢神经系统功能失常,严重威胁患儿生存质量[9,10]。基因学研究发现,SCN1A基因突变是导致Dravet综合征患者发病的重要原因,SCN1A基因所携带的遗传密码会指导组成钠离子通道的蛋白质的合成,进而改变脑部神经钠离子通道功能[11]。与此同时,SCN1A基因突变会影响钠离子在脑细胞间的流动,从而影响患儿细胞间电信号的正常传导[12]。大部分Dravet综合征患儿能检出SCN1A基因突变,通过MLPA法、Sanger测序检测点突变、大片段缺失/重复、小缺失、插入等方式对Dravet综合征患儿致病基因进行定位和评估[13]。统计数据发现,人体SCN1A基因主要定位在2号染色体长臂2q24.3,若其发生突变可导致钠离子的通道结构、功能出现异常,改变神经元兴奋性,诱发Dravet综合征发病[14,15]。相关研究显示,SCN1A基因突变为DS主要致病因素,但由于人种差异、样本量差异等相关因素影响,DS患儿的SCN1A基因突变发生率仍存在差异[16]。本研究发现,47例患儿中SCN1A基因突变阳性36例,突变发生率是76.60%,包括18例(50.00%)错义突变、18例(50.00%)截断突变;35例患儿是新生突变,1例患儿突变源自母亲,27例患儿的SCN1A基因突变是既往未提出新发突变,9例患儿的突变存在既往致病性报道;提示DS患儿存在高SCN1A基因突变发生率,临床可通过检测SCN1A基因突变发生率,为临床早期诊断DS提供信息。
研究表明,SCN1A基因突变的方式较为多样化,主要涉及剪切、错义、大片段缺失/重复、移码和无义等形式,其中错义突变可引起个别氨基酸变化,无义与移码突变能导致翻译终止密码子时间提前,并提前将蛋白质表达终止;而大片段缺失/重复、剪切突变则可引起碱基重排,造成转录、翻译异常,这种改变可引起钠离子通道快速失活功能异常,引起中枢神经系统神经元兴奋性增高、癫痫易感性增加,进而导致癫痫发作较为严重[17,18]。本研究通过分析SCN1A基因突变类型与Dravet综合征(DS)患儿临床特征的相关性发现,错义突变组DS患儿的不典型失神发作年龄、肌阵挛发作年龄均高于截断突变组DS患儿,但丛集样发作率低于截断突变组;错义突变组和截断突变组DS患儿的性别、首次发病年龄、惊厥持续状态发生率、癫痫发作类型、智力发育评估结果对比无显著差异;提示SCN1A基因突变类型和DS患儿的临床特征存在一定相关性[19,20]。而本研究发现,S4-S6区、非S4-S6区SCN1A基因错义突变患儿的首次发病年龄、不典型失神发作年龄、肌阵挛发作年龄、丛集样发作发生率、惊厥持续状态发生率、癫痫发作类型、智力评估结果对比无明显差异;考虑原因可能与基因型、临床特征异质性、纳入研究的样本量较少有关,后期可扩大样本量开展深入研究。
综上所述,Dravet综合征患儿存在高SCN1A基因突变发生率,且SCN1A基因突变类型与DS患儿不典型失神发作年龄、肌阵挛发作年龄、丛集样发作率等临床特征存在一定相关性。
猜你喜欢
昆明医科大学学报(2021年12期)2021-12-30
中老年保健(2021年11期)2021-08-22
眼科学报(2021年6期)2021-07-18
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
小天使·二年级语数英综合(2017年4期)2017-04-18
小天使·四年级语数英综合(2017年4期)2017-04-18
医学研究杂志(2015年12期)2015-06-10
少年文艺·开心阅读作文(2014年5期)2014-10-08
中学英语之友·中(2008年9期)2008-10-18
