
4-叠氮基-2,2,6,6-四硝基金刚烷的合成与热稳定性
2020-07-11 02:23:28蔡荣斌
含能材料 2020年7期
蔡荣斌,张 健,罗 军
(南京理工大学化工学院,江苏 南京 210094)
1 引言
由于含能材料内在的能量和稳定性的矛盾,兼具高能和钝感性质的高能量密度材料的研究一直是含能材料领域具有挑战性的前沿研究方向。金刚烷作为有机笼状高能量密度化合物的骨架母体之一,具有显著的优点,如:其结构刚性并高度对称,相对稳定;可官能化位点多,易于衍生;骨架上对称性地引入硝基、硝酰氧基、叠氮基、偕氟硝基等致爆官能团,将赋予其高能、耐热、钝感等优点[1],为解决含能材料能量和稳定性的矛盾提供了一种可能的方案。实际上,美国陆军武器研究发展和工程中心(AREDC)从20 世纪80 年代开始就注意到多硝基金刚烷,并将其列入到21 世纪发展战略中[2]。多硝基金刚烷的设计与合成一直吸引着有机化学家的注意,2,2-二硝基金刚烷[3]、1,2,2-三硝基金刚烷[4]、1,3,5,7-四硝基金刚烷[5]、2,2,6,6-四硝基金刚烷[3,6]、2,2,4,4-四硝基金刚烷[7-8]、2,2,4,4,6,6,6-六硝基金刚烷[9-11]等多硝基金刚烷相继被合成出来。此外,金刚烷碳骨架上同时引入硝基和硝酰氧基的4,4,8,8-四硝基金刚烷-2,6-二醇二硝酸酯[12]以及引入偕氟硝基的2,6-二氟-2,4,4,6-四硝基金刚烷和2,4,6,8-四氟-2,4,6,8-四硝基金刚烷[13]也于最近合成出来,本课题组还合成了一系列氮杂多硝基金刚烷[14-18]。
在金刚烷骨架上同时引入硝基与叠氮基尚未见报道,因此,本课题组将叠氮基团引入多硝基金刚烷骨架,设计了4-叠氮基-2,2,6,6-四硝基金刚烷(1)作为目标分子,考察叠氮基的引入对其热稳定性、密度和爆轰性能的影响,故在2,2,4,4,6,6,6-六硝基金刚烷合成[10]的基础上,对其进行逆合成分析(见图1)。
目标分子1 的偕二硝基可由二酮(2)的肟化、偕硝化引入,金刚烷骨架的叠氮基可由甲磺酸酯(3)的亲核取代反应得到,4 的分子内Aldol 反应关环构建金刚烷骨架3,甲酰基由双环壬烷二酮(5)的羰基α位单甲酰化引入,而5 可由Meerwein′s 酯(6)的水解脱酸来合成。通过逆合成分析,以丙二酸二乙酯及多聚甲醛为起始原料对目标分子4-叠氮基-2,2,6,6-四硝基金刚烷进行了合成研究,并利用核磁(1H NMR、13C NMR)、红外(IR)、元素分析(MS)等表征了相关中间体与目标分子1。用TG 和DSC 研究了4-叠氮基-2,2,6,6-四硝基金刚烷的热性能,并利用密度泛函理论计算了对4-叠氮基-2,2,6,6-四硝基金刚烷的密度、爆速和爆压。

图1 4-叠氮基-2,2,6,6-四硝基金刚烷的逆合成分析Fig.1 Retrosynthetic analysis of 4-azido-2,2,6,6-tetranitroadamantane
2 实验部分
2.1 合成路线
合成路线见Scheme 1,丙二酸二乙酯在N-甲基哌嗪催化下发生Knoevenagel 缩合,后在甲醇钠下环合,酸化得到Meerwein′s 酯(6),然后酸性水解脱羧得双环壬烷二酮5,酮α位单甲酰化得到醛4,L-脯氨酸催化醛4 分子内Aldol 缩合关环构建金刚烷醇7,甲烷磺酰氯(MsCl)对醇羟基保护得到甲烷磺酸酯3,然后用叠氮化钠亲核取代得到关键中间体叠氮化物2,最后经肟化,偕硝化合成目标化合物1。

Scheme 1 Synthetic route of 4-azido-2,2,6,6-tetranitroadamantane
2.2 仪器与试剂
Nicolet 傅里叶变换红外光谱仪(美国Thermofisher 公司);Bruker Avance. ⅡDRX 500MHz 核磁共振仪(德国Bruker 公司);Finnigan TSQ Quantumultra AM 型质谱仪(美国Thermal 公司);SDT Q600 DSC-TGA 同步热分析仪(美国TA 仪器公司)。
五氧化二氮参考[19]文献自制;丙二酸二乙酯,N-甲基哌嗪,甲醇钠,L-脯氨酸,甲酸乙酯,盐酸羟胺,无水硫酸钠均为分析纯,均出自阿拉丁;甲醇,乙醇,乙腈,二氯甲烷,N,N-二甲基甲酰胺,三乙胺等溶剂均为分析纯,均出自科隆试剂;盐酸,甲磺酰氯,叠氮化钠均为分析纯,均出自沪试。
2.3 合成实验
2.3.1 Meerwein′s 酯(6)的合成
参考文献[10]中的方法:将114 mL(0.75 mol)丙二酸二乙酯、18.75 g(0.625 mol)多聚甲醛、2.1 mL(0.019 mol)N-甲 基 哌 嗪 和100 mL 甲 苯 依 次 加 入500 mL 圆底烧瓶中,室温搅拌2 h 后升温100 ℃反应8 h,再加热到120 ℃反应10 h。减压蒸馏除去溶剂得到淡黄色粘稠液体,冷却至室温备用。将28.7 g(0.53 mol)甲醇钠溶于200 mL 无水甲醇,剧烈搅拌下快速加入上述淡黄色粘稠液体,室温搅拌反应30 min,70 ℃回流反应24 h,冷却至室温后,冰盐浴冷却下加入100 mL 乙醚,继续在冰浴冷却静置30 min。抽滤,滤饼用冰乙醚洗涤得到白色固体,将其溶于200 mL 蒸馏水中,用6 M 盐酸调节pH=4~5,析出大量白色固体,抽滤,滤饼用蒸馏水(200 mL)洗涤,干燥得51 g 淡粉色固体,收率70%。FT-IR(neat,ν/cm-1):2952,1737,1652,1442,1365,1330,1232;1H NMR(500 MHz,CDCl3)δ:12.18(s,2H),3.79(s,6H),3.78(s,6H),2.88(s,4H),2.34(s,2H);13C NMR(125 MHz,CDCl3)δ:172.49,171.93, 168.18, 96.91, 52.76, 51.94, 47.70,35.30,29.71。
2.3.2 双环[3.3.1]壬烷-2,6-二酮(5)的合成
参考文献[10]中的合成方法:将Meerwein′s 酯(6)(50.5 g,0.13 mol)和冰醋酸(120 mL)依次加入1 L 三口烧瓶中,加热至120 ℃使固体完全溶解,并缓慢向体系中滴加6 M 盐酸(82 mL,0.47 mol),滴加完全后继续反应12 h。冷却至室温,减压蒸馏除去溶剂,得浅黄色固体,固体用二氯甲烷(150 mL)和水(100 mL)溶解,取下层有机相,并依次用饱和碳酸氢钠水溶液(50 mL)、饱和食盐水(50 mL)洗涤,有机相用无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,得16.1 g 白色 固 体,收 率78%。FT-IR(neat,ν/cm-1):2935,2870,1694,1439;1H NMR(500 MHz,CDCl3)δ:2.71(d,J= 1.7 Hz,2H),2.61-2.52(m,2H),2.43-2.32(m,2H),2.18(s,2H),2.12-1.98(m,4H);13C NMR(125 MHz,CDCl3)δ:212.70,43.55,37.11,31.42,26.66。
2.3.3 2,6-二 氧 代 双 环[3.3.1]壬 烷-3-甲 醛(4)的合成
参考文献[10]中的合成方法:将化合物5(10 g,65.7 mmol)、甲酸乙酯(10.6 mL,131.4 mmol)和甲醇(75 mL)依次加入150 mL 的单口烧瓶中,冷却至0 ℃后缓慢滴加新配制的甲醇钠(4.26 g,78.8 mmol)的甲醇(10 mL)溶液。滴加完后,将体系升温至40 ℃搅拌反应12 h。待反应结束后,将反应体系冷却至室温后减压蒸馏除去溶剂,得黄色固体。将所得固体用二氯甲烷(150 mL)溶解,并用10% HCl 调节pH 至1~2,取下层有机相,并依次用饱和碳酸氢钠水溶液(100 mL)、饱和食盐水(50 mL)洗涤,有机相用无水硫酸钠干燥,过滤,减压蒸馏除去溶剂得黄色固体,硅胶柱层析,石油醚/乙酸乙酯(V∶V=5∶1)洗脱,得10.8 g 白色固体,收率92%。FT-IR(neat,ν/cm-1):2940,2850,1710,1650,1310,1240;1H NMR(500 MHz,CDCl3)δ:14.31(d,J= 1.8 Hz,1H),8.82(d,J=1.4 Hz,1H),2.84(m,1H),2.76-2.70(m,2H),2.45-2.40(m,2H),2.33-2.22(m,2H),2.07-2.06(m,3H);13C NMR(125 MHz,CDCl3)δ:212.25,189.58,183.28, 106.69, 43.58, 36.64, 34.92, 29.72,29.34,26.25 ;MS(EI):m/z(%)180(88)[M]+,152(48),123(32),105(38),95(66),77(52),55(100)。
2.3.4 4-羟基-2,6-金刚烷二酮(7)的合成
参考文献[10]中的合成方法:将化合物4(4.5 g,25 mmol)、L-脯氨酸(0.6 g,5 mmol)和乙腈(40 mL)依次加入100 mL 单口烧瓶中,在70 ℃反应8 h。反应结束后,冷却至室温,减压蒸馏除去乙腈,得黄色固体。将所得黄色固体用二氯甲烷(150 mL)和水(50 mL)溶解,取下层有机相,并用饱和食盐水(30 mL)洗涤,有机相用无水硫酸钠干燥,过滤,减压蒸馏除去溶剂得黄色固体,用二氯甲烷/正己烷重结晶,得4.2 g白色固体,收率94%。FT-IR(neat,ν/cm-1):3380,2940,2860,1700,1460,1290,1080;1H NMR(500 MHz,CDCl3)δ:4.36(m,1H),3.73(s,1H),2.81-2.79(m,2H),2.76-2.73,2.32-2.27(m,2H),2.22-2.12(m,2H),2.09-2.04(m,2H).13C NMR(125 MHz,CDCl3)δ:213.35,211.73,75.95,52.53,52.45,45.06,43.66,39.55,33.90,33.15。
2.3.5 4-甲磺酰氧基金刚烷-2,6-二酮(3)的合成
将化合物7(730 mg,4.05 mmol)溶于无水二氯甲烷(40 mL)中,在冰浴冷却及氮气保护下,加入无水三乙胺(1.25 mL,9 mmol),再缓慢滴加 甲磺酰氯(0.4 mL,4.98 mmol),继续反应2 h,升至室温反应16 h。加入饱和氯化铵溶液(20 mL)淬灭反应,分液,用二氯甲烷(20 mL×3)萃取水相,饱和食盐水洗涤有机相,经无水硫酸钠干燥,减压蒸馏得到橙黄色固体。硅胶柱层析,石油醚/乙酸乙酯(V∶V=1∶1~2∶3)洗脱,得到白色固体934 mg,收率89%。FT-IR(neat,ν/cm-1):3024,2937,2871,1717,1457,1336,1169,962;1H NMR(500 MHz,CDCl3)δ:5.19(td,J= 3.7,1.9 Hz,1H),3.14-3.06(m,2H),3.09(s,3H),2.72(m,J= 8.9,3.1 Hz,3H),2.37(dq,J= 13.5,3.1 Hz,1H),2.29(p,J= 3.1 Hz,1H),2.26(q,J= 3.0 Hz,1H),2.25-2.19(m,1H),2.16(dq,J=14.0,3.1 Hz,1H);13C NMR(125 MHz,CDCl3)δ:209.19,207.87,82.14,51.11,50.21,44.76,43.62,39.73,39.37,34.02,32.80;ESI-MS:m/z[(M+H)-]:259.00。
2.3.6 4-叠氮基金刚烷-2,6-二酮(2)的合成
将 化 合 物3(470 mg,1.82 mmol)和 叠 氮 化 钠(1.18 g,18.2 mmol)溶于无水DMF(20 mL)中,在氮气保护下,加热到120 ℃反应24 h。反应结束后冷却至室温,依次加入水(30 mL)和饱和碳酸氢钠(15 mL),用二氯甲烷萃取(25 mL×3),依次用饱和碳酸氢钠溶液、饱和食盐水洗涤有机相,无水硫酸钠干燥有机相后减压蒸馏除去溶剂得到淡黄色固体350 mg,收率94%。 FT-IR (neat,ν/cm-1):2942,2879,2114,1705,1452,1291,1227;1H NMR(500 MHz,CDCl3)δ:4.25(q,J=3.0 Hz,1H),2.92-2.83(m,2H),2.72-2.64(m,3H),2.34(dq,J=13.4,3.0 Hz,1H),2.27(tq,J=10.0,3.1 Hz,2H),2.24-2.08(m,2H);13C NMR(125 MHz,CDCl3)δ:209.73,209.45,66.93,49.86,49.64,44.87,43.93,39.71,34.66,34.14;ESI-MS:m/z[(M+Cl)-]:239.92。
2.3.7 4-叠氮基-2,2,6,6-四硝基金刚烷(1)的合成
化合物2(112 mg,0.54 mmol)、盐酸羟胺(150 mg,2.16 mmol)和无水乙酸钠(265 mg,3.23 mmol)溶于无水乙醇(10 mL),室温反应24 h。减压蒸馏除去溶剂,用乙酸乙酯(15 mL)和水(10 mL)溶解,分液,水相用乙酸乙酯萃取(5 mL×3),依次饱和碳酸氢钠、饱和食盐水洗涤有机相,干燥后减压蒸馏除去溶剂得到白色固体4-叠氮基金刚烷-2,6-二酮肟194 mg,未经纯化直接做下一步。
将上述白色固体194 mg、尿素(80 mg,1.32 mmol)、无水硫酸钠(3 g)加入到无水二氯甲烷(20 mL)中,氮气保护下,加热至回流,并在5 min 内滴加五氧化二氮(285 mg,2.64 mmol)的二氯甲烷(10 mL)溶液,反应液迅速变绿然后逐渐褪色至基本为无色。滴加完毕后继续反应30 min,冷却至室温,倒入到冷的饱和碳酸氢钠溶液(50 mL)中,用二氯甲烷萃取(20 mL×3),饱和食盐水洗涤有机相,无水硫酸钠干燥后减压蒸馏除去溶剂,得到的淡黄色固体进行硅胶柱层析,石油醚/乙酸乙酯(V∶V=1∶25~1∶20)洗脱得到白色固体49.3 mg,两 步 收 率25%。 FT-IR(neat,ν/cm-1):2967,2934,2927,2122,1574,1463,1300,1240;1H NMR(500 MHz,CDCl3)δ:4.09(q,J=2.7 Hz,1H),3.70(p,J=3.0 Hz,1H),3.49(p,J=2.7 Hz,1H),3.40(p,J=2.8 Hz,1H),3.34(p,J=2.8 Hz,1H),2.74(dq,J=15.6,3.2 Hz,1H),2.38(dt,J=15.9,3.2 Hz,1H),2.16(dq,J=15.6,3.3 Hz,1H),1.99-1.82(m,3H);13C NMR(125 MHz,CDCl3)δ:119.86,118.30,61.72,35.90,35.22,31.30,30.21,29.83,29.83,26.53;ESI-MS:m/z[(M+Cl)-]:392.10。
3 结果与讨论
3.1 合成路线分析
从Meerwein′s 酯(6)开始一直到4-羟基-2,6-金刚烷二酮(7)均参考本课题组优化过的合成工艺进行[10],结果均得到较高的产率,因此未进行进一步优化。
金刚烷骨架的叠氮基引入的方法主要包括:2-金刚烷酮在强酸如硫酸催化下与原位产生的氢氰酸亲核加成得到[20];或2-金刚烷醇在酸催化下与叠氮化钠发生SN1 反应引入叠氮基[21];或者大位阻喹啉磺酸保护的2-金刚烷醇与四叠氮化钛反应引入叠氮基[22]。当然,仲醇羟基发生Mitsunobu 反应[23-24]也是引入叠氮基的一种方法,但未见在金刚烷骨架上的报道。上述方法因为反应过程中存在产生剧毒易挥发的叠氮酸、或相关叠氮化试剂价格昂贵、或操作不便等缺点,因此探索更温和、安全、简便地在金刚烷骨架上引入叠氮基的方法具有重要的理论意义和实用价值。在本工作中设计了将金刚烷醇的羟基进行磺酰化再与叠氮化钠进行取代反应的策略将羟基转换为叠氮基。考虑到反应活性,采用空间位阻最小的甲磺酰氯对醇7 进行磺酰化得到甲磺酸酯3,为使反应顺利进行,选用了强极性非质子性的N,N-二甲基甲酰胺(DMF)作溶剂,并于120 ℃下反应,经过24 h 后反应完全,并以94%的高产率实现了4-叠氮基金刚烷-2,6-二酮的合成。
金刚烷骨架中引入偕二硝基的常用方法主要包括三种方法:第一种是酮肟直接用发烟硝酸进行偕二硝化反应[7];第二种是酮肟经过次卤酸氧化卤化、还原脱卤和氧化硝化三步反应[3];第三种是用绿色硝化试剂五氧化二氮对酮肟直接进行偕二硝化反应[8]。由于叠氮基不能容忍还原脱卤的反应条件,故并没有尝试第二种方法;首先探索了第一种方法,但未分离到目标产物;而利用绿色硝化试剂五氧化二氮对未分离的酮肟直接偕二硝化,以肟化、偕二硝化两步“一锅煮”合成了目标产物1,两步总收率为25%。
3.2 热稳定性
在 流 速 为50 mL·min-1的N2氛 围、升 温 速 率 为10 ℃·min-1、升 温 区 间 为50~500 ℃、样 品 量 为0.6200 mg 的条件下测试了4-叠氮基-2,2,6,6-四硝基金刚烷的TG-DSC 曲线,结果如图2 所示。由图2 可知,4-叠氮基-2,2,6,6-四硝基金刚烷自219 ℃样品开始质量损失,表明其具有较好的热稳定性;且在251 ℃有一个尖锐的放热峰,表明化合物1 在该温度下发生了剧烈的放热分解反应;至269 ℃样品质量损失约80%。
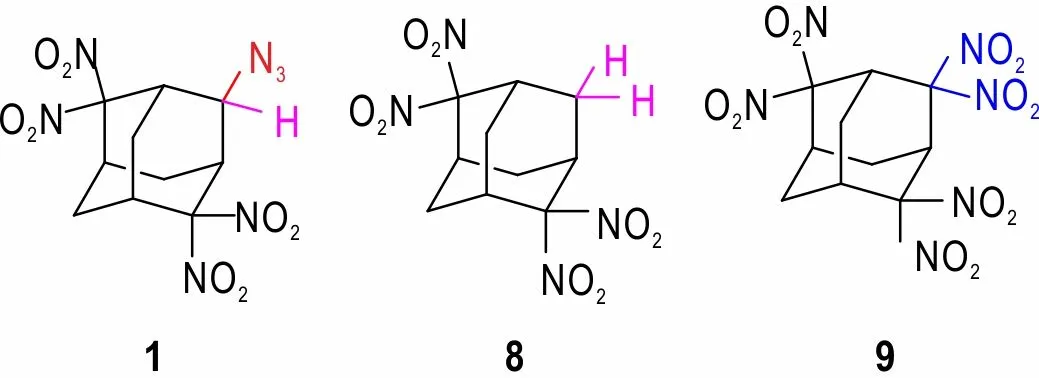
将化合物1 与其类似物2,2,6,6-四硝基金刚烷(8)[6]和2,2,4,4,6,6-六硝基金刚烷(9)[10]的热分析数据进行对比(见表1)。结果发现,与2,2,6,6-四硝基金刚烷(8)相比,化合物1 的初始分解温度(Tf)及分解峰温(Tp)均有所下降,说明,用叠氮基取代2,2,6,6-四硝基金刚烷4-位上的氢后,降低了结构的对称性,进而降低了目标物的热稳定性;与2,2,4,4,6,6-六硝基金刚烷(9)相比,化合物1 初始分解温度及分解峰温均大幅度提高,这是因为叠氮基取代2,2,4,4,6,6-六硝基金刚烷4-位上的偕二硝基后,降低了空间位阻,利于结构的稳定,进而提高了目标物的稳定性。

图2 化合物1 的TG-DSC 曲线Fig.2 TG-DSC curves of compound 1

表1 化合物1 与其类似物8 和9 的热稳定性Table 1 Thermal stability of compound 1 and its analogues(8 and 9)
3.3 爆轰性能的预估
采用密度泛函的B3LYP 基组计算了4-叠氮基-2,2,6,6-四硝基金刚烷(1)的密度、爆速和爆压,结果见表2。将2,2,6,6-四硝基金刚烷(8)和2,2,4,4,6,6-六硝基金刚烷(9)文献结果也列于表2,以便比较。
由表2 可见,4-叠氮基-2,2,6,6-四硝基金刚烷(1)的计算密度爆速、爆压均比(9)低,说明叠氮基取代2,2,4,4,6,6-六硝基金刚烷(9)4-位上的偕二硝基后,密度及爆轰性能均下降,这与预期结果一致;(1)的计算密度与(8)的密度[25]相差不大,但是爆速和爆压明显提高,说明叠氮基取代2,2,6,6-四硝基金刚烷4-位上的氢后后对密度影响不显著,但由于其为致爆官能团,因此可以明显提高爆速和爆压。

表2 化合物1 及其类似物8 和9 的密度及爆轰性能1)Table 2 Density and detonation properties of compound 1 and its analogues(8 and 9)
4 结论
(1)以丙二酸二乙酯和多聚甲醛为原料,经过环合、脱羧、甲酰化、Aldol缩合、O-甲磺酰化、叠氮化、肟化和偕硝化等步骤,合成了兼具硝基和叠氮基的新型笼形化 合 物4-叠 氮 基-2,2,6,6-四 硝 基 金 刚 烷,总 收 率9.8%。通过核磁共振、红外、质谱等对产物进行了表征。
(2)利用TG-DSC 对4-叠氮基-2,2,6,6-四硝基金刚烷的热性能进行了分析,结果为初始分解温度为219 ℃,峰温为251 ℃,表明其具有良好的热稳定性,有望用于钝感含能材料;此外,与2,2,6,6-四硝基金刚烷和2,2,4,4,6,6-六硝基金刚烷的热分析数据对比发现,用叠氮基取代2,2,6,6-四硝基金刚烷4-位上的氢后,会降低热稳定性,而用叠氮基取代偕二硝基后,会提高其稳定性。
(3)采用密度泛函理论预估4-叠氮基-2,2,6,6-四硝基金刚烷的密度为1.79 g·cm-3,爆速为7770 m·s-1,爆压为26.68 GPa。与2,2,4,4,6,6-六硝基金刚烷(9)相比的结果说明,叠氮基取代偕二硝基后,密度及爆轰性能均下降;与2,2,6,6-四硝基金刚烷(8)相比的结果说明,叠氮基的引入可以明显提高爆轰性能。
猜你喜欢
山西化工(2022年2期)2023-01-14 05:20:05
——以塔里木盆地塔中地区凝析油为例
石油实验地质(2021年5期)2021-11-01 06:55:44
地球科学与环境学报(2020年2期)2020-03-26 10:01:28
商品与质量(2019年32期)2019-11-29 05:56:00
山东冶金(2018年6期)2019-01-28 08:14:52
石油与天然气地质(2018年4期)2018-08-01 06:36:56
火工品(2018年1期)2018-05-03 02:27:56
中国资源综合利用(2017年3期)2018-01-22 02:45:40
合成化学(2015年9期)2016-01-17 08:57:14
山东工业技术(2015年1期)2015-03-16 01:27:53
