
Review of primary sclerosing cholangitis with increased IgG4 levels
2020-07-10 07:10CharisManganisRogerChapmanEmmaCulver
World Journal of Gastroenterology 2020年23期
Charis D Manganis, Roger W Chapman, Emma L Culver
Abstract
Key words: Primary sclerosing cholangitis; IgG4; IgG4-related disease; IgG4-related sclerosing cholangitis
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a chronic progressive disease characterised by fibrosis and strictures of intra-hepatic and extra-hepatic bile ducts, leading to cirrhosis, liver failure and eventual death[1]. It is classified as a rare condition, affecting less than 200000 individuals in the United States and 250000 individuals across the European Union. However, its incidence is rising, which has been linked to changing environmental exposures[2]. There is a strong association with inflammatory bowel disease (IBD), particularly ulcerative colitis (UC), present in 80% of patients[3,4]. Coexistent autoimmune conditions have been reported in up to 25%[5]. PSC is considered a pre-malignant condition, with an increased risk of hepatobiliary and colorectal carcinomas[6]. Currently, there is no medical treatment that improves survival, with liver transplantation the only option for cure[7].
Sub-types of PSC have been described, including classical large-duct PSC, smallduct PSC, and "overlap syndrome" with autoimmune hepatitis[8]. More recently, a subset of patients with PSC and high immunoglobulin G subclass 4 (IgG4) levels has been identified. Early evidence suggested that patients with PSC and high IgG4 levels may have a distinct clinical phenotype. Such patients need to be carefully distinguished from those with IgG4-related sclerosing cholangitis (IgG4-SC), the biliary component of multi-systemic IgG4-related disease.
In this review, we describe the diagnostic pathway, disease phenotype and associations, response to therapy and putative pathogenic mechanisms underlying PSC with high IgG4 levels. We propose that PSC can be clinically sub-classified by serum IgG4 levels, based on review of the literature, the evolution of clinical trial design in PSC to include serum IgG4 levels, and current clinical practice to risk stratify in tertiary high-volume centres diagnosing and managing patients with PSC.
LITERATURE SEARCH STRATEGY
We searched PubMed, MEDLINE and Embase for all articles with the search terms“primary sclerosing cholangitis”, “IgG4”, and “IgG4-related sclerosing cholangitis”:611 publications met the search criteria (536 in English language). Of these, 122 were review articles and 414 were original articles including case reports, case series,clinical cohort studies and clinical trials. We screened all abstracts and identified 108 articles with full text available relevant to this review; 36 of these specifically addressed primary sclerosing cholangitis and IgG4 levels. The abstracts and full text of relevant articles were independently assessed by two authors (EC and CM). A decision tree to reflect the literature search strategy is shown in Figure 1.
PREVALENCE OF PSC WITH HIGH IGG4 LEVELS
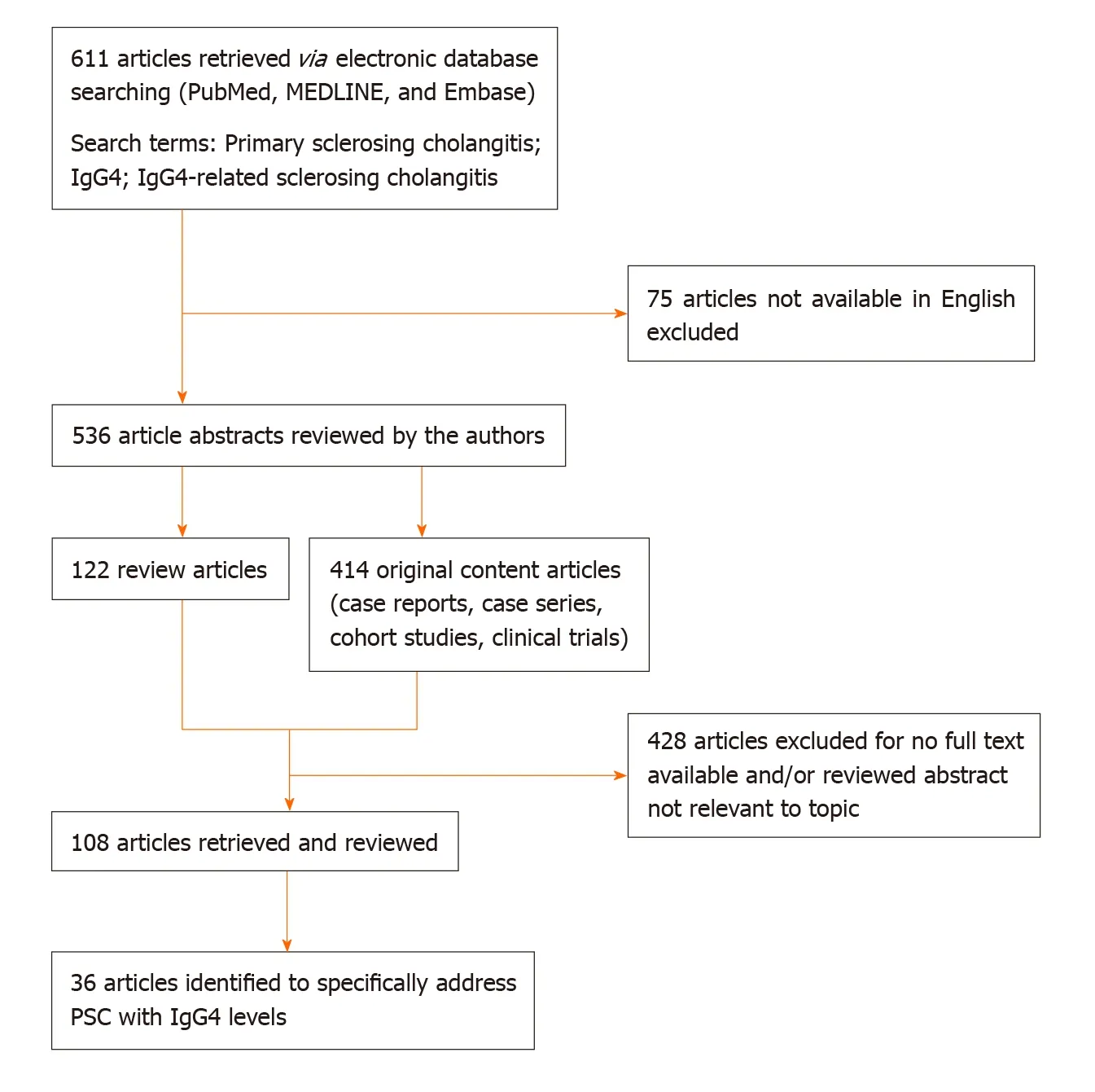
Figure 1 Decision tree for literature research strategy. PSC: Primary sclerosing cholangitis.
Early reports in the 1990’s describe patients with PSC-like biliary strictures and pancreatic disease, which was responsive to corticosteroid therapy[9-14]. In 2001,patients with biliary disease and a sclerosing autoimmune pancreatitis were reported to have elevated levels of serum IgG4 subclass (sIgG4). Mendeset al[15]reported elevated sIgG4 levels in 9% (12/127) of patients diagnosed with large duct PSC in 2006. Further retrospective studies including our own, have suggested that between 10% and 27% of patients diagnosed with PSC have an elevated sIgG4 level (PSC high sIgG4)[16-25](Table 1).
Case series describe patients diagnosed with PSC whose liver and/or bile duct tissue is infiltrated with abundant IgG4-positive plasma cells (> 10 per high power field) either diffusely or more focal in distribution[26]. Abundant IgG4-positive plasma cells have been described in 16% to 30% of liver explants in PSC patients at different stages of the disease (Table 2)[16,26-28]. Tissue infiltration by IgG4-positive plasma cells appears to correlate with sIgG4 levels, but this is influenced by a number of factors,such as the temporal association of biopsy and serum samples, and the introduction of immunosuppressive medications for co-existing inflammatory bowel disease and/or autoimmune conditions[16,26,27].
DIAGNOSIS OF PSC WITH HIGH IGG4 LEVELS
A diagnosis of classical large-duct PSC is made in the presence of cholestatic liver biochemistry, characteristic bile duct changes on cholangiography, and after wellknown secondary causes of cholangitis have been excluded[29]. The European Association for the Study of the Liver (EASL) Cholestatic Liver Disease Guidelines recommends that serum IgG4 levels are measured in all patients with large-duct PSC at diagnosis[29]. In those patients with an elevated sIgG4, it is important to distinguish PSC with high sIgG4 levels from IgG4-SC. If patients historically diagnosed with PSC and high sIgG4 levels are carefully categorised by clinical, radiological and histological means, between 4% and 12% may be re-classified as IgG4-SC in retrospective cohorts[16,19]. This has implications for management options, surveillance strategies and long-term prognosis.
Liver biopsy is not routinely required for the diagnosis of large-duct PSC, yet it may be indicated (1) to confirm small-duct PSC when there is clinical suspicion of cholestatic biochemistry and a normal cholangiogram; (2) to investigate evidence of autoimmune overlap with elevated liver enzymes, elevated titres of IgG and/orpositive autoantibodies (> 1/40 titres); and (3) to help diagnose IgG4-SC and IgG4-related hepatopathy in those with cholestatic biochemistry and elevated liver enzymes, and abnormal cholangiogram, and with (or without) elevated sIgG4 levels.Endoscopic retrograde cholangiopancreatogram (ERCP) is reserved for patients with PSC where therapeutic intervention is required (e.g., stricture dilatation or stenting),there is a high degree of suspicion for cancer (e.g., stricture sampling), or where MRCP views are suboptimal/un-obtainable[30]. However, biliary biopsies obtained at either ERCP or by direct-visualisation cholangioscopy can help to distinguish a dominant stricture due to advanced PSC, IgG4-SC or cholangiocarcinoma (CCA).There are currently no guidelines to recommend routine immunostaining of liver and/or bile duct biopsy specimens for IgG4-positive cells. However, this is performed in cases with a periductal fibrosis and predominant inflammatory (plasma cell)infiltrate in our centre, and in other tertiary referral centres for PSC and IgG4-SC in Europe.
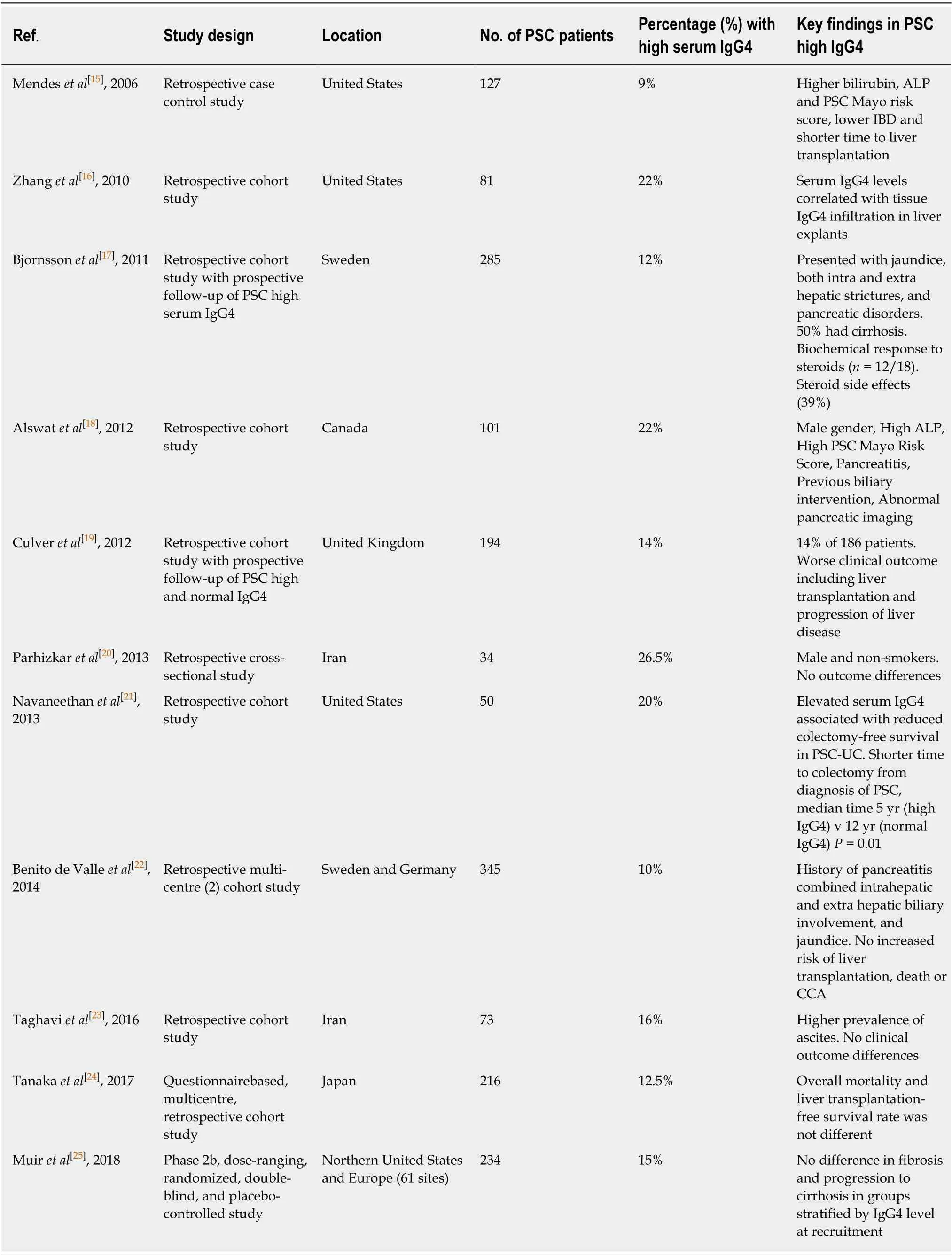
Table 1 Studies evaluating serum lgG4 in primary sclerosing cholangitis patients
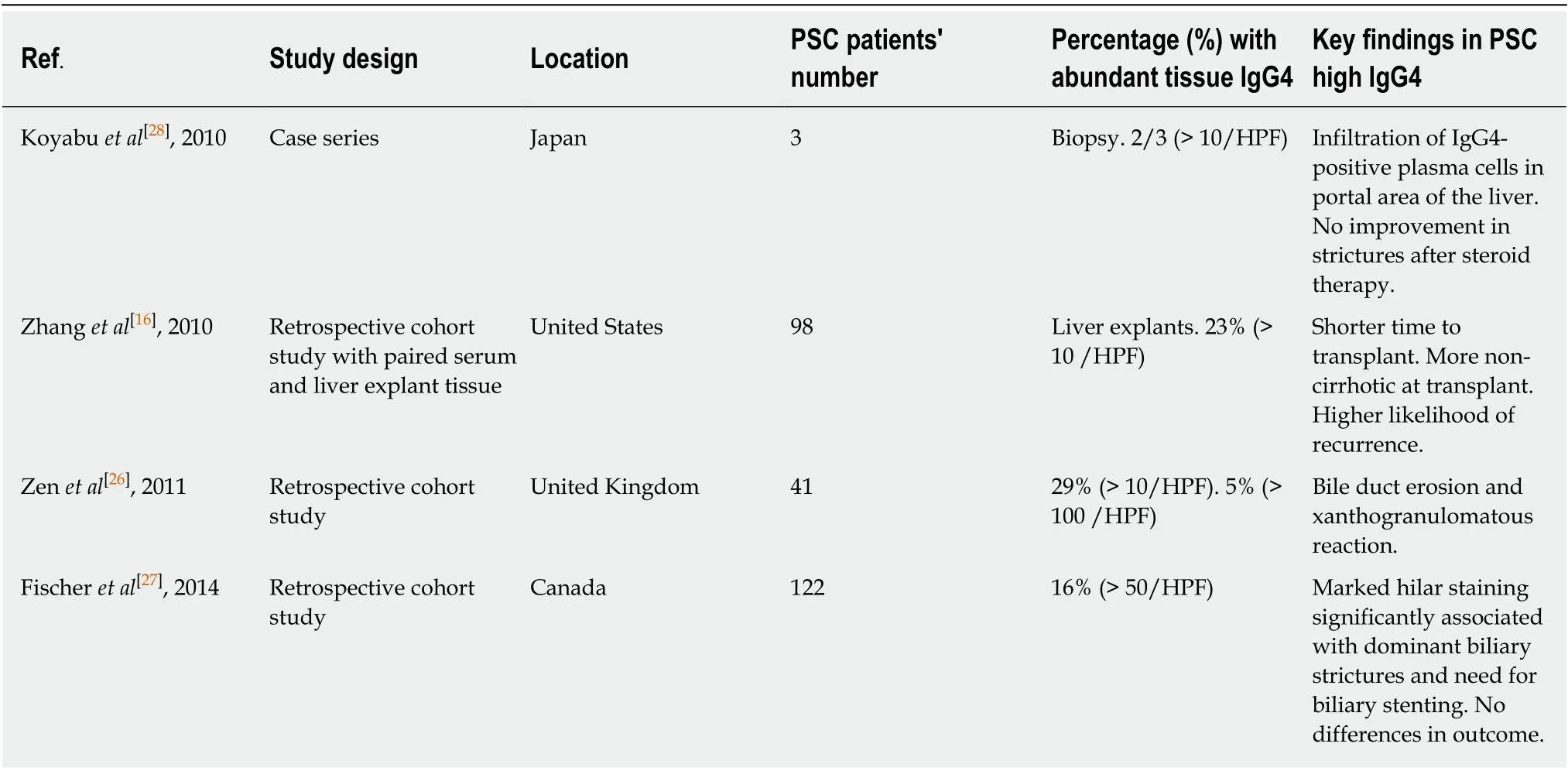
Table 2 Studies evaluating abundant tissue lgG4-positive plasma cells in primary sclerosing cholangitis patients
DIFFERENTIAL DIAGNOSIS OF SCLEROSING CHOLANGITIS WITH HIGH IGG4 LEVELS
IgG4-SC
IgG4-SC is the biliary manifestation of the multi-organ fibro-inflammatory IgG4-related disease (IgG4-RD). It has a male predominance, presenting in the 6thdecade,usually with obstructive jaundice, weight loss and abdominal discomfort. IBD is rarely seen [< 5% of cases with autoimmune pancreatitis (AIP) type 1], compared with over 80% prevalence of UC in large-duct PSC. IgG4-SC type 2 has intra-hepatic bile duct irregularity and strictures on cholangiogram that are challenging to differentiate from large duct PSC (Figure 2)[31,32]. Co-existent AIP type 1 is present in the majority of cases (> 90%) with involvement of the distal common bile duct[8]. Extra-pancreatic organ involvement is seen in over half of cases (e.g., bilateral sialadenitis,retroperitoneal fibrosis, aortitis, tubulointerstitial nephritis), which can be identified clinically and on imaging (CT chest, abdomen and pelvis, MRI of the head and neck,FDG-PET CT) if actively searched for.
Serum IgG4 levels are elevated in over 80%, serum IgE levels are elevated in 50%and a peripheral eosinophilia is present in 40% of IgG4-SC patients[33,34]. An elevated serum IgG4 level over four times the upper limit of normal has a high specificity (>98%) to distinguish IgG4-SC from PSC, but a low sensitivity and positive predictive value[34,35]. Elevated serum IgG4 levels over two times the upper limit at diagnosis has been associated with multi-organ involvement and disease relapse in those with IgG4-SC[34]. A ratio of serum IgG1 to IgG4 of > 0.24 in those with an elevated serum IgG4 has been validated in two cohorts (Dutch and United Kingdom) to distinguish PSC high sIgG4 from IgG4-SC with high specificity (> 98%)[19,34,35]. Elevated serum IgE levels (> 125 kUL) and peripheral eosinophilia are more prominent in IgG4-SC than PSC, and historic series that document PSC patients with a peripheral eosinophilia and corticosteroid responsiveness may indeed have IgG4-SC[33,36-40]. Complement C3 and C4 levels can be low in IgG4-SC/AIP, especially in those with IgG4-related renal disease, and is rarely found in PSC high sIgG4; p-ANCA does not have a good discriminatory value[41]. More recently, a novel IgG4:IgG RNA ratio has been shown to provide excellent discrimination between IgG4-SC, PSC and CCA, regardless of the sIgG4 level, and now require real-life validation.
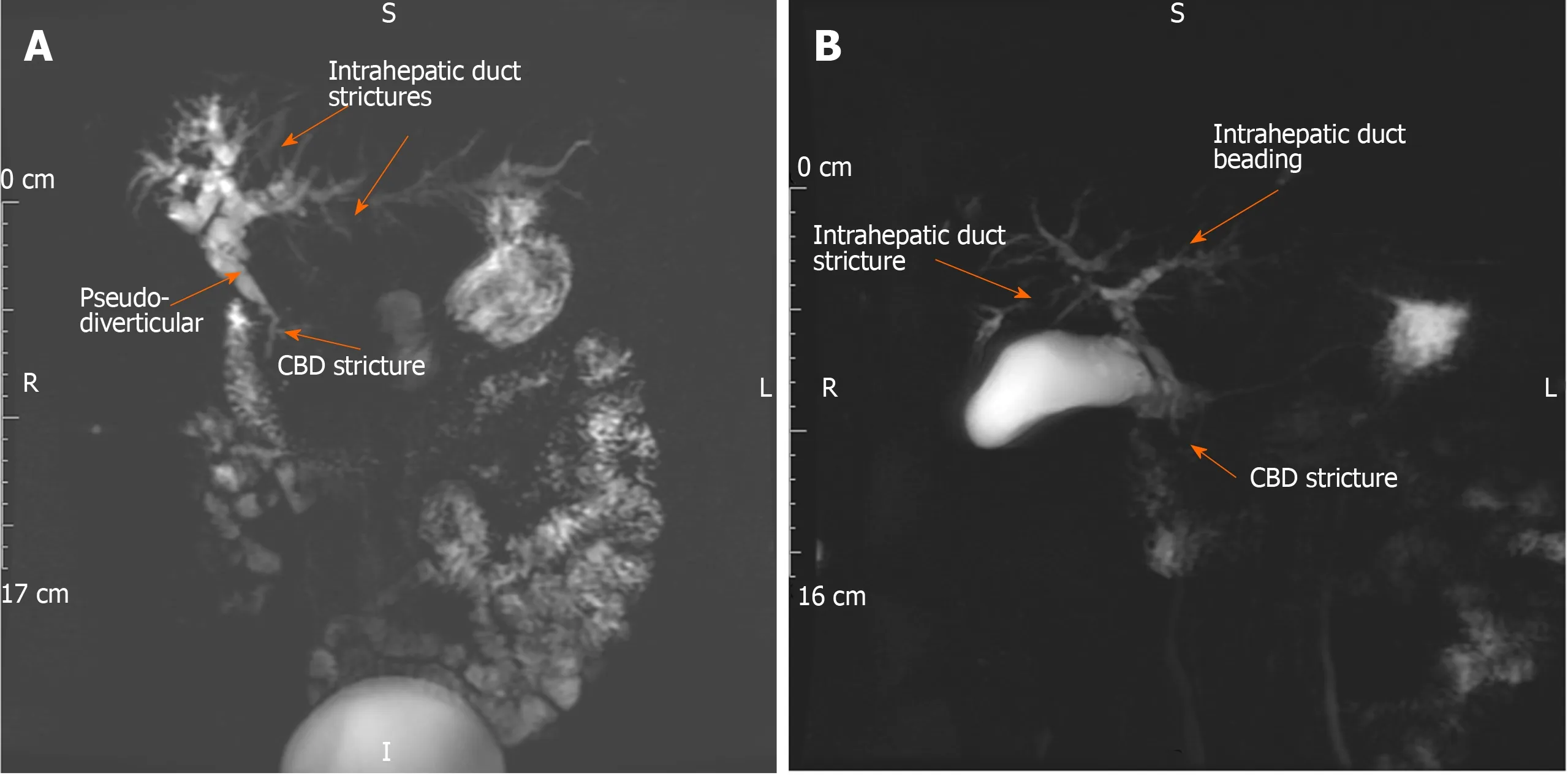
Figure 2 lgG4-related sclerosing cholangitis type 2 (primary sclerosing cholangitis-like). A 58-year old female with cholestatic liver biochemistry, elevated serum IgG4 and IgE, and peripheral blood eosinophilia. A: Magnetic resonance cholangiopancreatography (MRCP) demonstrates common bile duct stricture (CBD)(arrow), intrahepatic duct irregularity and strictures (arrows) and beading (arrows). The liver biochemistry, serum IgG4 level, common bile duct and intrahepatic duct strictures improved after 12 wk of corticosteroid therapy. Endoscopic retrograde cholangiopancreatography and brushings of the CBD stricture were negative for dysplasia. B: MRCP demonstrates new right intra-hepatic duct stricture (arrow) and improvement of CBD stricture (arrow) one year later. Azathioprine (1.5 mg/kg)started to reduce further risk of disease relapse. CBD: Common bile duct.
The histological hallmarks of IgG4-RD are a lymphoplasmacytic infiltrate with abundant IgG4-positive plasma cells, storiform pattern of fibrosis, obliterative phlebitis and variable presence of eosinophils in affected organs[42]. Liver and bile duct biopsy morphological findings can be inconclusive; ”storiform fibrosis” and”obliterative phlebitis” is not always seen in small samples in IgG4-SC, and an”onion-skin” fibrosis (peri-ductal concentric fibrosis) is not exclusive to PSC[43].However, if present then they help to support the diagnosis. Abundant IgG4-positive plasma cells (> 10 per high power field in biopsy; > 50 per high power field in resection specimens) and a ratio of IgG4 to IgG-positive plasma cells > 40% in the context of classical morphological features are favourable for a diagnosis of IgG4-SC rather than PSC high IgG4[42].
Diagnostic criteria for IgG4-SC such as the Mayo HISORt for IgG4-SC (Histology,Immunology, Serology, Other organ involvement, Response to corticosteroid therapy)can be used to support a diagnosis (Table 3)[44]. A clinical scoring system has been proposed to distinguish between IgG4-SC type 2 and PSC with intra-hepatic strictures based on age, other organ involvement and beading on cholangiogram, reporting excellent discrimination (area under the receiver operating curve 0.99) between IgG4-SC and PSC in a single-centre cohort of 39 IgG4-SC and 76 PSC patients[45]. However, a mid-range score (5-6 points) prompts a diagnostic steroid trial to distinguish the two conditions, and the extent to which more advanced IgG4-SC or PSC high sIgG4 itself responds to corticosteroids is uncertain.
Based on our experience as a high-volume tertiary referral centre for PSC and IgG4-SC we have included a list of features to hep distinguish the two conditions (Table 4)[1,7,15,18,19,27,29,32,36,41,42,44-67]. Ultimately, appropriate diagnosis of PSC is crucial for optimising surveillance for disease progression and hepatic decompensation, the need for liver transplantation and malignant complications. Identification of IgG4-SC is important, as early introduction of immunosuppression improves clinical symptoms,cholestatic liver biochemistry, bile duct strictures/wall thickening on cholangiogram and may prevent advancement of disease and organ damage in both the liver and other involved organs.
Secondary sclerosing cholangitis
Sclerosing cholangitis can be secondary to a number of infections, vascular incidents,infiltrative conditions, immunological insults, toxins, traumatic insults and congenital disease. We have shown that serum IgG4 can be elevated in a variety of infective andinflammatory conditions[19]. It is often less than twice the upper limit of normal and falls with correction of the underlying cause[34]. Clinical history should guide the further investigation and management in this setting.
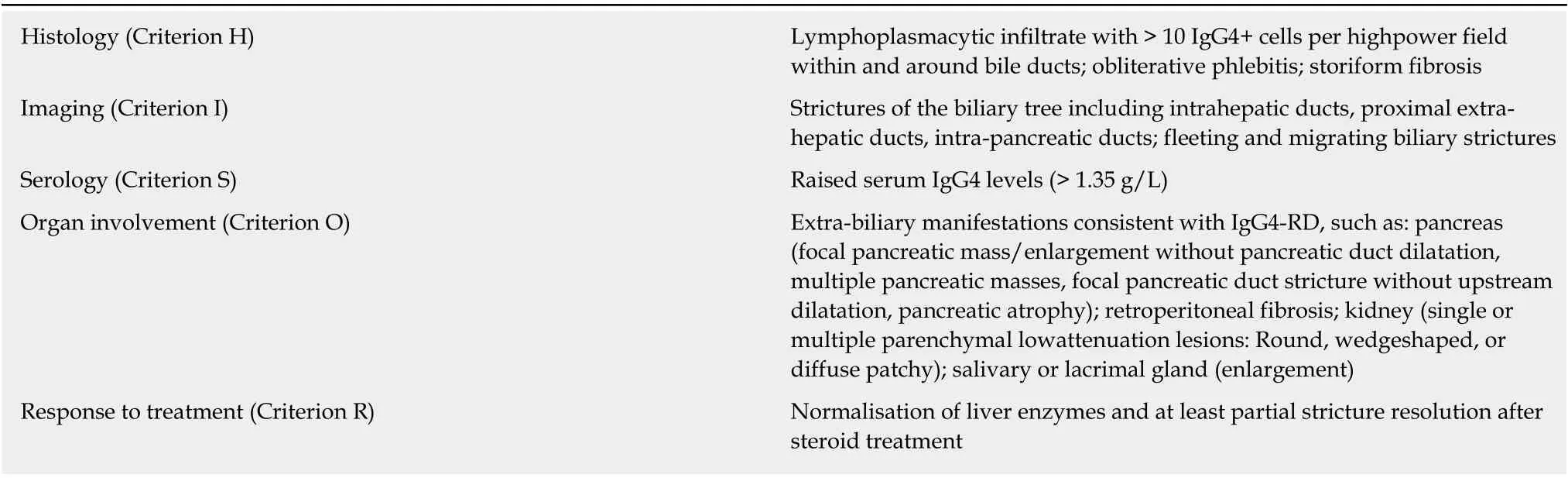
Table 3 HlSORt Criteria for lgG4-related sclerosing cholangitis (Adapted from references[32,44])
CCA
PSC with a dominant stricture and IgG4-SC type 1, 3 and 4 can be difficult to distinguish from a CCA. CCA usually presents with cholestasis, obstructive symptoms and a dominant stricture in the hilum, intra or extra-hepatic bile ducts with upstream dilatation. An elevated sIgG4 has been reported in up to 20% of patients with extra-hepatic CCA, although usually between one and two times the upper limit[34]. An elevated sIgG4 level over four times the upper limit has a high specificity(100%) to distinguish between IgG4-SC and CCA, but case reports of CCA arising on the background of IgG4-SC exist, and PSC is a pre-malignant condition, so sampling should always be done to rule out malignancy[6,7,29,34]. Similarly, the IgG4:IgG RNA ratio has been reported to provide excellent discrimination between IgG4-SC, PSC and CCA, but requires real-life prospective validation[35]. CA 19-9 can be elevated in obstructive jaundice independent of the cause and is not a useful discriminator[68].Corticosteroids may reduce the peri-tumoral inflammatory response even in CCA,and so a trial may not be discriminatory. CCA needs to be excluded in all cases as a priority, when it may be amenable to curative surgical therapy.
Abundant IgG4-positive cells have been described in resection specimens in the peri-tumoral tissue of CCA, located around the periphery of dysplastic foci[69].Morphological assessment of tissue is required to distinguish a background of PSC or IgG4-SC, although biliary biopsies from ERCP and/or cholangioscopy are often small so can be inconclusive. Brushings are acquired for evidence of dysplasia. One small study reported that the concentration of IgG4 in bile fluid obtained at ERCP could distinguish PSC high IgG4, IgG4-SC and CCA (n= 54), but this has yet to be reproduced in other series[70,71].
CLINICAL DISEASE PHENOTYPE OF PSC WITH HIGH IGG4 LEVELS
The clinical characteristics of patients with PSC and high IgG4 levels have been described in a number of retrospective studies. The Mayo Clinic first reported that patients with PSC high sIgG4 (n= 12/127) had a higher level of total bilirubin and alkaline phosphatase (ALP), and a higher PSC Mayo Risk Score, than those with normal IgG4 levels[15]. A follow-on study of 285 PSC patients, carefully characterised 24 PSC high sIgG4 patients from the point of sIgG4 measurement; one-half presented with jaundice, one-fifth had pancreatic disease, all had both intra- and extra-hepatic duct strictures, and one-half had cirrhosis[17]. A study of two independent European cohorts with 345 PSC patients from Germany and Sweden similarly reported an elevated sIgG4 level was associated with a prior history of pancreatitis, combined intra- and extra hepatic duct involvement and jaundice in multi-variate analysis[22]. In a Canadian cohort of “all-comers with sclerosing cholangitis” (n= 101), an elevated sIgG4 was associated with elevated ALP, liver enzymes and PSC Mayo Risk Score[18].The caveat to interpretation to these studies is that none accounted for the duration of PSC or prior/current immunosuppressive treatment for associated colitis; the latter can confound IgG4 measurements[34].

Table 4 Features to distinguish primary sclerosing cholangitis with high serum lgG4 from lgG4-related sclerosing cholangitis
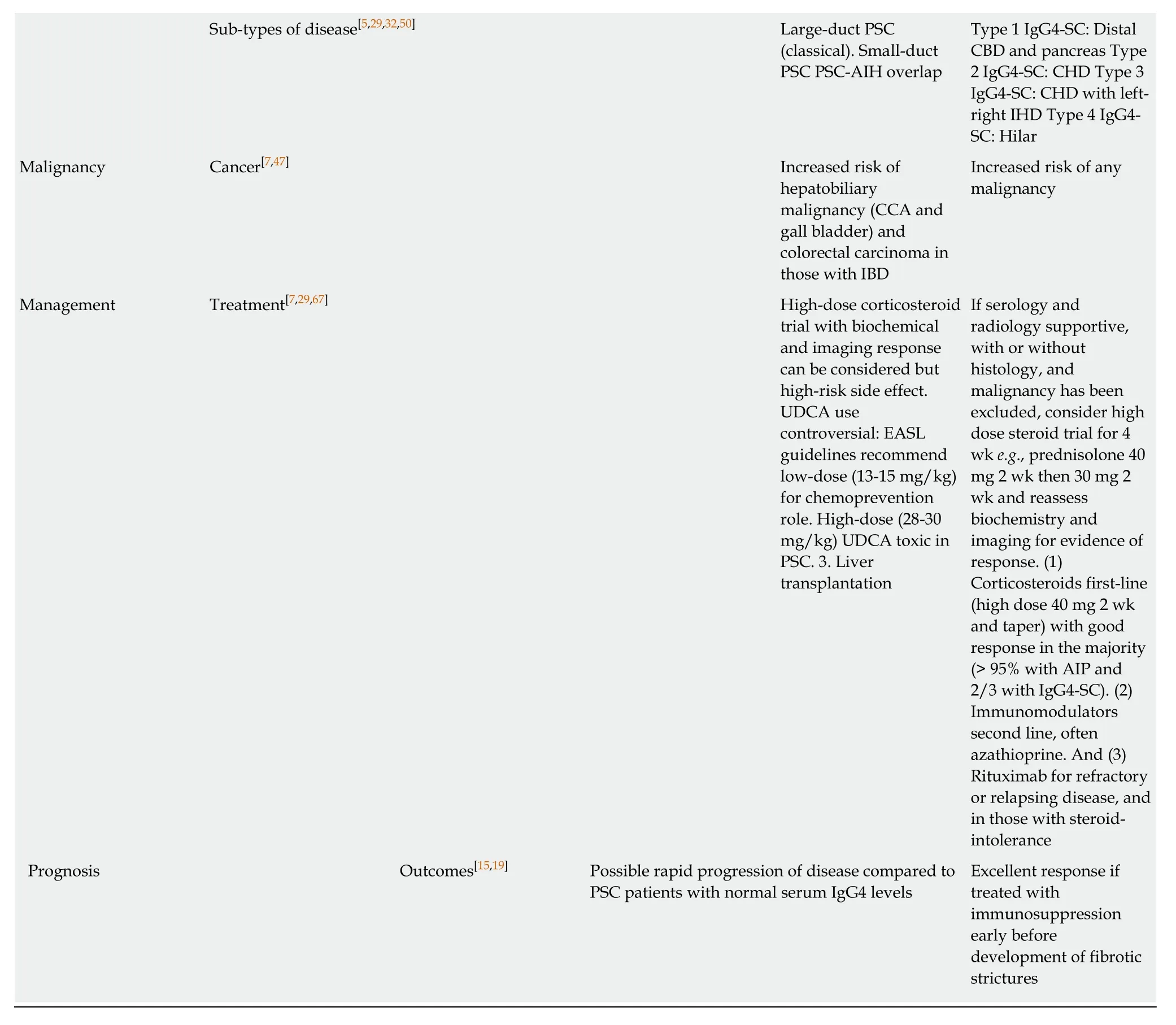
AIP: Autoimmune pancreatitis; ALP: Alkaline phosphatase; CBD: Common bile duct; CCA: Cholangiocarcinom; EASL: European Association for the Study of the Liver; IBD: Inflammatory bowel disease; PSC: Primary sclerosing cholangitis; UC: Ulcerative colitis; IgG4-SC: IgG4-related sclerosing cholangitis;MRCP: Magnetic resonance cholangiopancreatography; HLA: Human leukocyte antigen; UDCA: Urseodeoxycholic acid.
Histological assessment in liver explants of advanced PSC has demonstrated that patients with PSC and abundant IgG4 infiltration have differences in distribution and severity of biliary strictures compared with those with minimal IgG4 infiltrates.Marked hilar IgG4-positive infiltration (> 50/HPF) was associated with dominant strictures and need for biliary stenting in one study[27].
COLITIS IN PSC AND HIGH IGG4 LEVELS
PSC has strong links with IBD, with a reported association in Northern Europe and America of 60%-80% with UC[3,4]. The colitis of PSC-IBD has a unique clinical phenotype: a pan-colitis with prominent right sided inflammation, back-wash ileitis,rectal sparing, a quiescent and prolonged sub-clinical course.
Retrospective studies have reported a reduced frequency and more aggressive colitis in PSC with high IgG4 levels compared to normal IgG4 levels. Mendeset al[15]reported less frequent IBD (50%vs85%) in those with PSC and high sIgG4 versus normal sIgG4 levels. Navaneethanet al[21]reported that those patients with PSC-UC and high sIgG4 levels were younger at PSC diagnosis, more likely to have endoscopic and histological evidence of backwash ileitis, have a flare of colitis in the preceding five-years, have a reduced colectomy-free survival and a shorter time to colectomy (5 yearsvs12 years) after twelve-year median follow-up than those with normal sIgG4 levels.
Abundant IgG4 plasma cell infiltration in the colon (> 10 per HPF) has been reported in up to 10% (8/76) of PSC-UC patients, with an IgG4 to total IgG ratio of less than 40%[19]. We, and others, have shown that a higher IgG4 plasma cell count in the colon correlates with colitis severity in those with UC, independent of PSC diagnosis[62]. In contrast, IgG4-SC is rarely associated with an inflammatory colitis (<5% of patients) and the colon is often infiltrated with > 50/HPF IgG4 positive plasma cells but no other morphological features.
MALIGNANCY IN PSC AND HIGH IGG4 LEVELS
Patients with PSC have an increased risk of hepatobiliary carcinoma, particularly CCA and gallbladder carcinoma, and in those with PSC-UC (and to a lesser extent in PSC-Crohn’s colitis) an increased risk of colorectal dysplasia and cancer[6]. Studies on PSC and high IgG4 levels have been underpowered to assess the risk of malignancy in this cohort. There were no differences in the rates of CCA, colorectal cancer and colorectal dysplasia in one small study of PSC-UC high IgG4 (n= 10)vsnormal IgG4(n= 40) over a ten-year follow-up period[21]. There are no studies directly addressing surveillance strategies for hepatobiliary or colorectal malignancy according to IgG4 levels in PSC, and therefore current surveillance in this cohort follows the national guidelines for PSC[72].
There is an independent association of the presence of IgG4 antibodies and malignancy; acting as a mechanism for immune evasion in malignant melanoma and abundant in the peri-tumoral tissue in pancreatic carcinoma and CCA[73,74].Furthermore, IgG4-RD itself has been associated with an increased risk of malignancy in both retrospective and prospective cohort studies, especially within the first year of diagnosis, and often away from sites of involvement in the disease[69]. However, there is currently a lack of prospective controlled data to truly define this risk.
THERAPEUTIC OPTIONS FOR PSC AND HIGH IGG4 LEVELS
There is no current medical therapy that improves survival in patients diagnosed with PSC. Ursodeoxycholic acid improves liver biochemistry, but there is no evidence it alters histological progression, time to liver transplantation or mortality, whilst high doses (28-30 mg/kg) are considered toxic with an increased risk of malignancy[75,76].Liver transplantation is currently the only option for progressive disease.
Corticosteroids
Several small early studies have evaluated the use of systemic and endo-biliary corticosteroid therapy in patients with PSC, with some showing evidence of clinical and biochemical improvement but concerns being raised about toxicity and disease progression[77-82]. In a case series of three patients with PSC high sIgG4 treated with high-dose corticosteroids, there was biochemical improvement but no reversal of strictures and two had rapid disease progression[28]. In a cohort study, 18 of 24 PSC high sIgG4 patients were treated with oral corticosteroids (alongside biliary stenting),and two-thirds had a biochemical response, but no reversal of strictures was reported[17]. Of concern, high rates of adverse events were reported in the steroid group (39%) including hypoglycaemia, diabetes, osteopenia and psychological disturbance. Furthermore, the group were heterogeneous, with 2 of the 18 diagnosed with AIP thus likely had IgG4-SC, and 1 of the 18 diagnosed with a CCA. The 6 who did not receive steroids had contraindications (n= 1) or only mild elevations of liver biochemistry (n= 5).
Paediatric patients who present with features of large-duct PSC on cholangiogram and/or small-duct PSC on liver biopsy will often have autoimmune features(associated with an inflammatory liver infiltrate in up to 90% of patients), which are highly responsive to corticosteroid treatment, dubbed “autoimmune sclerosing cholangitis”[83]. We have demonstrated in a small paediatric series that a quarter of these have an elevated serum IgG4 and/or infiltration of IgG4-positive plasma cells in the liver tissue[84]. It has been hypothesised that in these PSC patients with prominent plasma cell infiltrate, there is an opportunity to halt the active inflammatory element,which may benefit from steroids and possibly further immunomodulation. Adult PSC patients with autoimmune hepatitis overlap, some with inflammatory colitis, and an eosinophilia often respond favourably to steroids[72]. Those that benefit tend to be younger with a shorter disease duration, and therefore are less likely to have "burntout" disease.
Immunosuppressive therapies
Patients with PSC-IBD and/or co-existent autoimmune diseases may be on a second or third-line immunosuppressive agent. Case reports and case series have reported favourable improvements in cholestatic liver biochemistry and liver histology in patients with PSC on azathioprine, even in the absence of autoimmune hepatitis overlap[81]. Small trials have also shown improvements in liver biochemistry with tacrolimus, mycophenolate and methotrexate, and in liver histology with cyclosporine, although these have not specifically determined IgG4 level[85-88].Concomitant use of these agents for IBD, AIH-overlap and other autoimmune disease has made it difficult to assess their effect on PSC alone per se[89].
Biological agents
A recent multi-centre randomised placebo-controlled phase 2b study of Simtuzimab, a monoclonal antibody directed against LOX-L2 that aims to prevent collagen crosslinking, stratified PSC patients according to serum IgG4 level. The study was negative in its primary outcome to detect mean change in hepatic collagen content. In subanalysis, improvement in fibrosis was more common in the simtuzumab-treated groups among patients with elevated sIgG4 (> 140 mg/dL) at baseline[25]. This may be explained by the recently reported role of B cells and plasmablasts driving fibrosis in patients with IgG4-RD, which may also apply to the PSC high sIgG4 subtype[25,90]. This theory will require further exploration.
Antibodies to TNF-alpha are used in patients with IBD, and mechanistic studies have suggested that TNF-alpha may have a pathogenic role in PSC[91]. Whilst retrospective series suggested some benefits in ALP levels in PSC, a double-blind randomised controlled study showed no beneficial effect of infliximab and was stopped early due to futility, with a suggestion of harm[92]. In a retrospective PSC-IBD cohort from the Mayo treated with monoclonal antibodies (infliximabn= 42,adalimumabn= 19), only adalimumab was associated with a significant decrease in ALP at 6-8 mo, which was not sustained at 12-24 mo, and there was no radiological improvement[93]. However, to date anti-TNF response has not been stratified by IgG4 levels.
B-cell depletion with Rituximab is efficacious in patients with IgG4-SC, currently advocated for disease relapse, immunosuppressive failure and steroid-intolerance[67].A case report in a patient with PSC and co-existent rheumatoid arthritis suggested improved liver enzyme profile, and stable cholangiographic appearances[94]. In an uncontrolled retrospective study of patients after liver transplantation, the addition of rituximab to the regimen was reported to delay PSC recurrence in the graft[95].
Case series and observational cohorts have assessed anti-integrin therapies such as vedolizumab in PSC-IBD, with conflicting results (comprehensively summarised by Lynchet al[96]). Whilst some patients had a fall in ALP, others had a rise in ALP and ALT levels, independent of their endoscopic/histological colitis activity response[96].None were stratified by sIgG4 level.
Novel therapies
There has been a steady increase in trials of novel therapeutic agents for PSC, with the dual aims of understanding disease pathogenesis and delaying the progression of disease. There are currently multiple agents under investigation, including bile acid mimetics (nor-UDCA), Farnesoid X receptor agonists, apical sodium-dependent bile acid transporter inhibitors, antimicrobials such as vancomycin and anti-VAP1 monoclonal antibodies[97]. For the most part, small numbers of patients, inadequate characterisation, and differing endpoints have limited sub-group analysis of PSC high IgG4.
OUTCOME IN PSC WITH HIGH IGG4 LEVELS
The prognosis of untreated PSC is poor, with estimations of transplant-free life expectancy ranging from 12-21 years from diagnosis[98,99]. It has been proposed that disease outcome can be stratified by a number of variables, including age at presentation, serum ALP, the extent of biliary involvement, presence of cirrhosis and sIgG4 levels[48,100-102].
Early evidence suggested that PSC high sIgG4 had a worse clinical outcome than PSC normal sIgG4. Mendeset al[15]reported patients with PSC high IgG4 had a reduced time to liver transplantation (1.7 yearsvs6.5 years), although overall survival was unchanged. Bjornssonet al[17]similarly reported PSC high IgG4 had a higher risk of progression to cirrhosis compared with those with normal levels. We also found an increased risk of progression of liver disease and requirement for liver transplantation in the group with PSC high sIgG4 levels[19]. Furthermore, in PSC patients having undergone liver transplantation, abundant IgG4 liver infiltrates were associated with a more aggressive clinical course, shorter time to transplantation, more non-cirrhotic patients at the time of transplant and an increased risk of recurrence[16]. However,none of these studies accounted for the duration of PSC and in some, failure to identify IgG4-SC may have delayed appropriate immunomodulatory treatment and favoured fibrotic progression, skewing observed results.
Larger studies have failed to demonstrate an impact of sIgG4 on outcome. In a Japanese PSC cohort (n= 216), high sIgG4 levels at diagnosis were associated with a higher rate of liver transplant, but this association was not preserved when controlling for confounding variables[24]. In a large European two-centre PSC cohort (n= 345), there was no adverse impact on outcome measures of liver transplant, liverrelated death or cancer development[22]. In the randomised placebo-controlled study of Simtuzumab, the PSC high sIgG4 group did not show a more fibrotic phenotype or increased progression of cirrhosis over the 96 wk trial[25]. Other smaller retrospective studies have also failed to show a significant impact of sIgG4 levels on outcome[22,24,28].One study showed abundant tissue IgG4-positive infiltration did not impact on outcome measures, although interpretation of these results is limited as low numbers of IgG4-positive cells were considered as abundant[27].
PATHOGENIC MECHANISMS IN PSC HIGH IGG4
PSC is considered to be an immune-mediated disease. Several mechanisms have been proposed, involving dysregulation of immune signalling, increased permeability of the bowel wall allowing exposure of toxins to the liver, and damage from toxic bile acids (for a comprehensive review see Karlsenet al[2]). An intriguing question is how and if PSC high IgG4 differs in its pathogenesis from IgG4-SC and PSC with normal IgG4 levels (Figure 3).
GENETIC ASSOCIATIONS
Various risk loci in genome-wide associations (GWAS) and Immunochip studies in PSC have been identified, particularly representing the major histocompatibility complex (MHC) on chromosome 6. Human leukocyte antigen (HLA) DRB1*0301,DRB1*1301 and DRB1*1501, associated with autoimmune disease, have been identified as associated with PSC, although no single susceptibility gene is known[2,54-57]. A recent genetic analysis of HLA haplotypes in PSC highlighted different associations in patients with PSC with high and normal sIgG4 levels (n= 263)[58]. HLAB*08 allele (strongest PSC risk factor) was less prevalent in patients with PSC high sIgG4 (> 1.35 g/L and > 2.0 g/L), whereas HLA-B*07 and DRB1*15 (on the same conserved haplotype, AH7.1) were more prevalent in PSC high sIgG4 (IgG4 > 2.0 g/L)[58]. This finding was replicated in Swedish and US cohorts[58]. A recent GWAS in 857 IgG4-RD patients in Japan and over 2000 healthy controls identified the HLADRB1 and FCGR2B regions as susceptibility loci for IgG4-RD[103]. Asian studies have reported higher frequencies of the DRB1*0405-DQB1*0401 haplotype in Japanese patients with AIP compared to healthy individuals and chronic calcifying pancreatitis[59]. In the United Kingdom, we have demonstrated an increased frequency of HLA-DRB1*0301-DQB1*0201 in a multi-centre cohort of IgG4-SC/AIP patients compared to healthy individuals[60]. Thus, whilst there are overlaps in HLA class II DRB1 associations in PSC (high and normal sIgG4) and IgG4-SC, certain haplotypes are more prominent in the PSC high IgG4 subtype (HLA-B*07 and DRB1*15).
IGG4 ANTIBODIES
IgG4 is the least prevalent of the four IgG subclasses in health, representing 3%-6% of total IgG. Physiological IgG4 responses can be induced by repeat or high antigen exposure and are associated with tolerance induction such as in immunotherapy.IgG4 production is driven predominantly by T helper-2 cytokines (IL4 and IL13)which induce IgG4 and IgE antibodies, T-regulatory cytokine (IL10) and Tfollicular/peripheral helper cytokine (IL21) that shifts this balance towards IgG4,known as the “modified Th2 response”[104,105]. The glycosylation pattern of an antibody can influence its cellular functions and pathogenicity, including complement and Fc receptor binding. We demonstrated that glycosylation of the Fab arms of IgG4 antibodies is increased compared to that of other IgG subclasses[106]. We showed altered IgG4 Fab and IgG1 and IgG4 Fc antibody glycosylation in IgG4-SC and PSC high sIgG4; with disease-specific enhancement of IgG4-specific Fab sialylation,reduction in IgG1-specific Fc bisection and recovery with steroid treatment in IgG4-SC, and reduced IgG1-specific sialylation in those with PSC[106]. Ultimately, the prominence of IgG4 antibodies in some patients with PSC and IgG4-SC may result in differential response to antigen exposure and downstream Fc-effector functions compared with those with normal IgG4 levels.
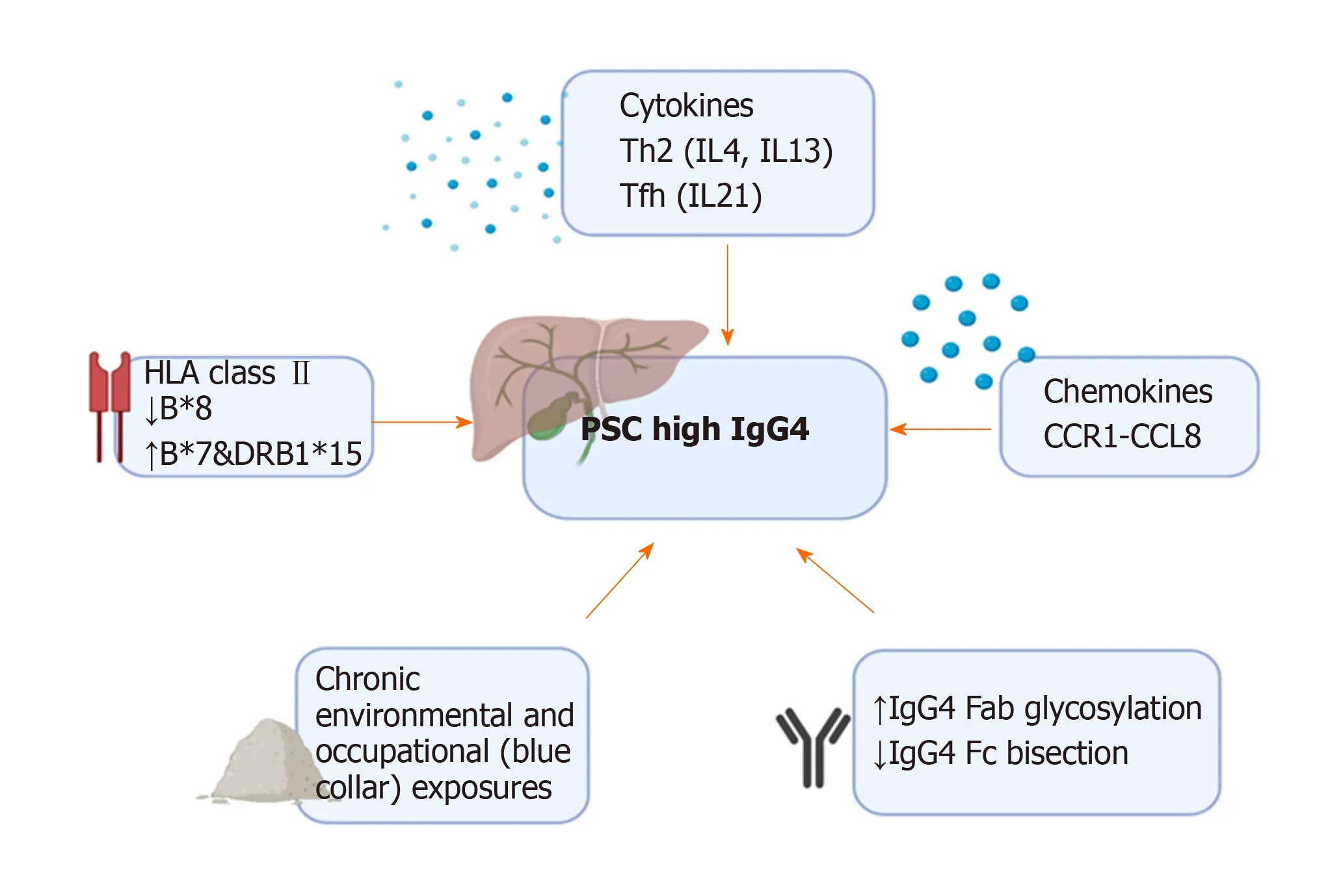
Figure 3 Pathogenic factors driving the phenotype of primary sclerosing cholangitis with an increased lgG4 level. HLA: Human leukocyte antigen; PSC: Primary sclerosing cholangitis.
CHRONIC ANTIGEN EXPOSURE
IgG4 can protect against IgE-mediated allergic reactions, and it has been speculated that the chronically raised IgG4 levels may represent chronic activation of this protective mechanism in the presence of long-term antigen exposure[107]. We described chronic exposure to environmental antigens, such as occupational exposures in “blue collar workers”, more frequently in those with IgG4-SC compared with all PSC patients, and in PSC high sIgG4 compared with normal sIgG4 levels[108]. We also demonstrated a polyclonal IgG4 response to diverse antigens in IgG4-SC and PSC high sIgG4 compared with PSC normal IgG4 and heathy controls, supporting expansion of pre-existing IgG4-switched B cells in both conditions[109].
IMMUNE ENVIRONMENT
Chemokines recruit inflammatory cells to affected tissues, and differences in chemokine and cytokine expression have been demonstrated in the liver of patients with IgG4-SC, PSC high and normal sIgG4 levels. In IgG4-SC and PSC high IgG4 there is over-expression of CCL1 (highly expressed in peri-biliary epithelium), CCR8(important in recruitment of Th2 and T-regulatory cells), and cytokines IL4 and IL10,compared with PSC normal IgG4[110]. CCL1 chemotactic factor was positive in infiltrating lymphocytes in PSC high IgG4 compared with peri-biliary glands,pancreatic duct epithelium, and vascular endothelial cells involved in obliterative phlebitis affected in IgG4-SC[110]. Thus, elevated IgG4 levels in PSC (and IgG4-SC) may represent a sustained immunoreaction to unidentified antigen(s), and favour a type-2 cytokine response, derived from Th-2 and T-regulatory cells.
CONCLUSION
PSC patients with high IgG4 levels appear to have a distinct clinical phenotype, HLA associations, cytokine and chemokine profiles, and post-translational antibody modifications compared to PSC patients with normal IgG4 levels. It is important to distinguish PSC high IgG4 from IgG4-SC, other causes of secondary sclerosing cholangitis and CCA - all of which can have elevated IgG4 levels. Corticosteroids may improve biochemical parameters in all conditions, however there are concerns regarding steroid toxicity and disease progression in a sub-group of patients with PSC high IgG4 and more advanced cirrhosis. Currently, lack of robust data, an absence of well-designed prospective studies, and the aforementioned safety concerns limit acceptability and use of immunosuppressants in the context of PSC high IgG4. This may be more applicable in paediatric variants with “autoimmune sclerosing cholangitis” and overlap with an autoimmune hepatitis with predominant IgG4-positive plasma cells. Moving forward, we believe it will be important to continue to incorporate the IgG4 level into prospective clinical trial design of novel therapies in PSC.
 World Journal of Gastroenterology2020年23期
World Journal of Gastroenterology2020年23期
- World Journal of Gastroenterology的其它文章
- Liver-directed therapies for liver metastases from neuroendocrine neoplasms: Can laser ablation play any role?
- Potential of the ellagic acid-derived gut microbiota metabolite - Urolithin A in gastrointestinal protection
- Endosonographic diagnosis of advanced neoplasia in intraductal papillary mucinous neoplasms
- Medications in type-2 diabetics and their association with liver fibrosis
- Pancreatic necrosis and severity are independent risk factors for pancreatic endocrine insufficiency after acute pancreatitis: A long-term follow-up study
- Impact of a national basic skills in colonoscopy course on trainee performance: An interrupted time series analysis