
大疱性系统性红斑狼疮二例
2020-07-04 02:07:00陈显侠余炳前郑佳媛牛桃香李一凡骆志成
中国麻风皮肤病杂志 2020年7期
陈显侠 余炳前 郑佳媛 牛桃香 李一凡 骆志成
兰州大学第二医院皮肤科,甘肃兰州,730030
大疱性系统性红斑狼疮(bullous systemic lupus erythematosus,BSLE)是一种罕见的自身免疫性水疱性疾病,其发病率为0.2/106~0.5/106[1], 常好发于20~40岁的中年女性。现报道2例典型的大疱性系统性红斑狼疮,并对相关文献进行复习。
临床资料例1,女,36岁。因“躯干、四肢红斑、水疱伴痒半年”于2019年6月20日入院。患者半年前无明显诱因躯干、四肢散在红斑,部分有水疱,在当地医院就诊,诊疗不详,皮损反复发作,日晒后加重。半月前日晒后皮损加重,自觉瘙痒明显,伴口腔溃疡、脱发、双手指关节痛,否认雷诺现象。在当地医院就诊,查“ds-DNA抗体弱阳性、抗核抗体弱阳性、抗Sm抗体阳性、抗SSA抗体阳性、抗1- SNRNP抗体阳性;补体C3、C4降低;血沉40 mm/h;皮肤组织病理检查:表皮角化不全,有中性粒细胞浸润,基底细胞液化变性,真皮浅层见薄层均质化黏蛋白沉滞(图1);直接免疫荧光检査:IgA、lgG、C3呈线状沉积于基底膜带(图2,3),诊断为“疱疹样皮炎”,给予“甲泼尼龙12 mg,日2次”等治疗7天,患者上述症状未见明显改善,遂住院治疗。既往否认传染病史、药物食物过敏史;家族中无类似疾病史。体检:生命体征平稳,心肺腹未査及明显异常,躯干、四肢可见散在及密集分布的大小不一、形状不规则的红斑,其上可见结痂,部分红斑上可见绿豆至蚕豆大水疱,疱壁厚,疱液澄清,尼氏征阴性,部分水疱破溃,可见糜烂面(图4);双手指间关节轻压痛,无肿胀;口腔黏膜未见损害。入院后辅助检査:血常规示白细胞计数8.75×109/L,中性粒细胞比率0.80,淋巴细胞比率0.15,嗜酸性粒细胞比率0.00,红细胞计数3.96×1012/L,血红蛋白124 g/L,血小板222×109/L;补体C3 0.48(0.60~1.60)g/L;抗核抗体阳性1∶320;抗Sm抗体阳性;抗SS-A抗体阳性;胸部CT:双侧腋窝淋巴结增大;心脏彩超:心内结构未见异常,左室功能正常,肺动脉压正常范围,二尖瓣反流(轻度)、三尖瓣反流(轻度);余检查结果无明显异常。诊断:BSLE。给予甲泼尼龙40 mg/d静滴;硫酸羟氯喹片0.2g,日2次;卤米松软膏、复方多黏菌素软膏外用等治疗11天,患者无新出皮损,红斑颜色较前明显变淡,水疱干涸结痂,瘙痒明显缓解,出院后随访,至今病情平稳无复发。
例2,女,19岁。因“全身出红斑、水疱伴痒痛2个月”于2019年6月25日入院。患者2个月前无明显诱因舌体出单个米粒大水疱,未予重视,后水疱破溃形成糜烂面,且面部、躯干、四肢逐渐出现散在红斑、水疱,自觉痒痛,伴口腔溃疡、脱发、间断发热(体温未测)、关节肿痛、口干、眼干等症状,否认雷诺现象、光敏感。在外院就诊,皮肤组织病理检查:表皮下水疱,真皮乳头部显著炎细胞浸润,中性粒细胞灶性聚集(图5);直接免疫荧光检查:基底膜带IgG、IgA、C3呈带状沉积(图6、7),诊断为“大疱性类天疱疮?大疱性红斑狼疮?线状IgA大疱性皮病?”,给予“醋酸泼尼松25 mg日1次”等治疗5天,疗效欠佳,遂住院治疗。既往否认传染病史、药物食物过敏史;家族中无类似疾病史。体检:生命体征平稳,心肺腹未查及明显异常,额部、躯干、四肢可见散在分布的大小不一、形状不规则的红斑,部分上有水疱,疱壁紧张,尼氏征阴性,部分水疱沿红斑边缘呈环形排列(图8);口腔黏膜未见损害。入院后辅助检查:血常规示白细胞计数3.9×109/L,中性粒细胞比率0.48,血红蛋白99 g/L;粪常规:隐血试验阳性;24 h尿蛋白定量0.22 g/24h(0~0.15);抗核抗体阳性1∶320;抗SS-A抗体阳性、抗SS-B抗体阳性、Ro-52抗体阳性;腹部B超示:脾大、脾静脉增宽,肝胆、胰、双肾未见异常;余检查未见异常。诊断:BSLE。给予醋酸泼尼松片20 mg日2次;卤米松软膏、复方多粘菌素软膏外用等治疗5天,原水疱大部分干涸,右手有一个新出水疱,激素加量为醋酸泼尼松片25 mg日2次;并联用硫酸羟氯喹片0.2 g日2次治疗,出院后随访,至今病情平稳无复发。
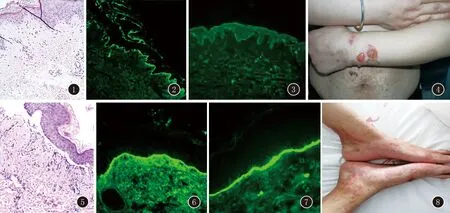
图1表皮角化不全,有中性粒细胞浸润,基底细胞液化变性,真皮浅层见薄层均质化黏蛋白沉滞(HE,×200)图2、3IgA、lgG、C3呈线状沉积于基底膜带(免疫荧光,×200)图4左上肢红斑、水疱,疱液澄清,尼氏征阴性图5表皮下水疱,真皮乳头部中性粒细胞浸润(HE,×200)图6、7IgA、lgG、C3呈带状沉积于基底膜带(免疫荧光,×400)图8双足红斑、水疱,尼氏征阴性
讨论BSLE是发生在系统性红斑狼疮(SLE)患者的获得性表皮下水疱性疾病,临床罕见,仅占SLE发病率的0.4%[2]。SLE患者中有75%会出现皮肤损害,其中极少数会出现大疱性皮损,但并非所有伴大疱性皮损的SLE均称为BSLE。通常SLE的水疱和大疱可出现在以下三种情况:①界面空泡性皮炎;②SLE合并其他自身免疫性水疱性疾病(如大疱性类天疱疮);③与VII型胶原蛋白抗体相关的自身免疫性水疱性疾病,最后一种情况是BSLE[3,4]。BSLE大疱性皮损可发生于SLE病程的任何阶段,而且,它的出现一般与病情活动度和内脏损害有关,可合并严重的肾脏损害或神经系统受累[3,5]。
1993年Gammon等[6]总结了BSLE诊断要点,而Yell等[7]1995年进一步修改了此诊断标准,建议符合以下3项条件即可诊断为BSLE:①符合SLE的诊断标准; ②并发表皮下水疱或大疱;③DIF或IIF检查发现 IgG 和(或)IgM、IgA沉积或结合于BMZ。由于BSLE临床罕见,容易误诊,其主要与SLE合并原发性水疱性疾病,如大疱性类天疱疮(BP)、线状IgA大疱性皮病(LABD)、疱疹样皮炎(DH)、获得性大疱性表皮松解症(EBA)等相鉴别。组织病理方面,BSLE表现为表皮下水疱,伴大量中性粒细胞浸润,真皮浅层大量黏蛋白沉积,相比之下,DH和LABD除了有中性粒细胞外,还有嗜酸粒细胞浸润,而BP患者炎性细胞以嗜酸粒细胞为主,EBA则缺少炎性细胞。免疫病理
方面,根据免疫蛋白类型、浓度及沉积部位可相互区分,例如:BSLE几乎所有类型的免疫球蛋白均在基底膜带呈颗粒状或线状沉积,EBA患者IgA和IgM数量较BSLE少,BP仅存在IgG和C3蛋白沉积,LABD为IgA呈线状沉积,DH患者的IgA呈颗粒状沉积于真皮乳头。除此之外,ELISA法检测血清中VII型胶原蛋白抗体及对IgG抗体行亚组分析,这项新型检测法对BSLE的诊断及鉴别诊断也具有积极的指导作用[8]。
BSLE的治疗上,由于其临床表现的特殊性,目前尚未发现其特效治疗。传统治疗SLE的糖皮质激素仅对少数BSLE患者有效,而使用氨苯砜治疗(2 mg/kg·d)的患者大多数都能取得很好的疗效,甚至较小的剂量(25~50 mg)都会有明显的治疗效果,但也不能忽视其带来的毒性作用。除此之外,甲氨蝶呤、硫唑嘌呤、环磷酰胺等免疫抑制剂也对BSLE的治疗有一定效果[8,9]。近年来,越来越多研究表明生物制剂,尤其利妥昔单抗,其作为CD20嵌合单克隆抗体,可降低BSLE患者VII型胶原蛋白抗体的浓度,从而对难治性BSLE具有显著疗效,这有望成为日后研究BSLE治疗的重点[10,11]。
本报道中的两例患者均符合SLE的诊断标准,结合皮损特点、组织病理及免疫病理可明确诊断为BSLE,只因条件有限,无法完善BP180、BP230、VII型胶原抗体等检查。由于BSLE临床罕见,例1曾被误诊为疱疹样皮炎,例2入院时初步诊断为线状IgA大疱性皮病,完善自身抗体后明确诊断为BSLE,因此对于发生在中青年女性的大疱性疾病,应该积极完善自身抗体、免疫球蛋白等相关检查。
猜你喜欢
山东冶金(2022年2期)2022-08-08 01:50:44
山东冶金(2019年1期)2019-03-30 01:34:54
兽医导刊(2016年12期)2016-05-17 03:51:42
兽医导刊(2016年12期)2016-05-17 03:51:36
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:17:01
山东青年(2016年2期)2016-02-28 14:25:33
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:36
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:33
应用化工(2014年10期)2014-08-16 13:11:29
养殖与饲料(2014年10期)2014-02-28 22:14:55
