
基于体细胞突变数据库筛选免疫原性结直肠癌高频新抗原的方法
2020-06-29 01:21秦丽丽李毅坚梁兆端陈蕾李文慧陈超黄亚灵张乐刘松明邱思葛玉萍彭文婷林欣欣张秀清董旋李波
遗传 2020年6期
秦丽丽,李毅坚,梁兆端,陈蕾,李文慧,陈超,3,4,黄亚灵,张乐,3,4,刘松明,3,4,邱思,4,葛玉萍,彭文婷,3,林欣欣,4,张秀清,4,董旋,李波,4
技术与方法
基于体细胞突变数据库筛选免疫原性结直肠癌高频新抗原的方法
秦丽丽1,2,李毅坚1,梁兆端1,陈蕾1,李文慧1,陈超1,3,4,黄亚灵1,张乐1,3,4,刘松明1,3,4,邱思1,4,葛玉萍1,彭文婷1,3,林欣欣1,4,张秀清1,4,董旋1,李波1,4
1. 深圳华大生命科学研究院,深圳 518083 2. 大理大学基础医学院,大理 671000 3. 中国科学院大学华大教育中心,深圳 518083 4. 武汉华大吉诺因生物科技有限公司,武汉 430079
结直肠癌是世界高发和高致死率的恶性肿瘤。靶向新抗原的免疫治疗已被证实可以诱导癌症患者肿瘤持续消退,但这些特异性新抗原,仅适用于个体精准治疗。随着大量的高频肿瘤基因突变被发现,这些与突变相关的高频新抗原可覆盖更多人群,具有较强的临床意义。然而目前结直肠癌中是否也存在高频新抗原仍不清楚。本研究利用来源于321个结直肠癌患者的体细胞突变数据库,联合1种标准过滤和7种预测算法,筛选并获得了25个基于中国人高频分型HLA-A*1101限制性的高频新抗原,它们均具有高亲和力(IC50<50 nmol/L)和高呈递分值(>0.90);其中,除了阳性对照多肽KRAS_G12V8-16外,11个高频新抗原能够在体外诱导细胞毒性T淋巴细胞(cytotoxic T lymphocyte, CTL)分泌γ干扰素(interferon gamma, IFN-γ),证实具有免疫原性。选取免疫原性最强的新抗原C1orf170_S418G413-421及阳性对照多肽KRAS_G12V8-16体外刺激T细胞,利用流式细胞分选及单细胞转录组测序技术,获得其特异性CTL的免疫组库信息,所构建的TCR-T(T-cell receptor engineered T cell)能够识别新抗原并分泌细胞因子。以上结果表明,本研究开发了一种利用体细胞数据库预测并体外筛选验证具有免疫原性高频新抗原的方法,为结直肠癌及其他癌种的多肽、DC(dendritic cells)疫苗、TCR-like抗体、TCR-T等免疫治疗提供了重要的多肽靶点和TCR信息,具有实际的临床应用价值。
结直肠癌;高频;新抗原;呈递
结直肠癌(colorectal cancer,CRC)是目前全球范围内第2致死和第3高发的恶性肿瘤[1]。传统的外科手术、化疗、放疗以及靶向治疗等方法,均不能有效提升患者的5年生存率[2]。近年来,免疫治疗作为新兴有效的肿瘤免疫疗法,已经被广泛应用在结直肠癌的治疗中[3]。过表达亲和力增强的T细胞受体(T-cell receptor,TCR)或者基于抗体的人工嵌合抗原受体(chimeric antigen receptor, CAR)工程化改造T细胞,使其靶向肿瘤相关抗原(tumor associated antigens, TAAs),如癌胚抗原CEA[4,5]和人表皮生长因子HER2[6],虽然能够抑制结直肠癌的转移,但也会导致患者产生严重的自身性免疫反应。因此,发现肿瘤特异性新抗原,对于提高结直肠癌过继性T细胞疗法的有效性和安全性具有重要价值。
肿瘤特异性抗原(tumor-specific antigens, TSAs)由肿瘤细胞蛋白编码区的体细胞突变产生,与在正常细胞中低表达但在肿瘤细胞中高表达的TAAs相比[4~6],仅存在于肿瘤细胞中[7]。在肿瘤中,累积的突变包括非同义、无义、插入/删除(InDel)和移码,又可分为驱动突变(driver mutations)和乘客突变(passage mutations),前者多与细胞生长失控和肿瘤转移有关,后者不影响致瘤表型但会增加免疫原性[8]。突变的新抗原多肽由主要组织相容性复合物(major histocompatibility complex, MHC)分子呈递到肿瘤细胞表面,然后被T细胞识别,从而导致强烈的抗肿瘤作用[9]。高度特异的新抗原可实现个性化的癌症免疫疗法,但限制了其广谱性的使用范围[10]。数据显示,一些肿瘤由于微卫星不稳定而导致高肿瘤突变负荷(tumor mutational burden, TMB)进而常会产生一些高频的新抗原[10]。在结直肠癌患者中,约15%~20%的患者具有微卫星不稳定,并且结直肠癌的缺陷错配修复(deficient mismatch repair, dMMR)具有远高于标准值的TMB[3],预示结直肠癌可产生大量高频新抗原。此外在两种类型的突变中,驱动突变仅占有8%的CD8+T细胞新表位,而乘客突变则产生92%的CD8+T细胞新表位和100% CD4+T细胞新表位[8]。同时,在递呈多肽的HLA分子中,HLA-A*1101等位基因是美国白种人、亚裔美国人和中国人[11]高频分型(http://www.allelefrequencies.net/)。因此,找到靶向两种类型突变并受HLA-A*1101限制的高频新抗原,不但可以提高结直肠癌免疫疗法的疗效、安全性和适用率,还可以降低结直肠癌临床治疗的成本和时间,具有重要的临床实际意义。本研究建立了一套分析与验证的方法,从321例结直肠癌患者的体细胞突变数据中预测了HLA-A* 1101限制的高频新抗原,并验证了它们的免疫原性,为结直肠癌免疫治疗的提供了新的靶点,具有较强的临床应用价值。
1 材料与方法
1.1 实验材料
抗原处理相关转运蛋白(transporters associated with antigen processing, TAP)缺陷的T2细胞系(CRL-1992)和HEK-293细胞系(CRL-1573)均购自美国ATCC中心。采用含10%Hyclone胎牛血清(Healthcare公司,美国)的Gibco IMDM培养基(Thermo Fisher公司,美国)培养T2细胞,以及含10%胎牛血清的Gibco DMEM培养基(Thermo Fisher公司, 美国)培养HEK293细胞。过表达HLA-A*1101分子的慢病毒感染T2细胞系后,对细胞进行HLA分型鉴定,确认获得HLA-A*1101分型的T2细胞系。采集已签署知情同意书的健康自愿者的外周血,通过Ficoll-Hypaque梯度离心法分离外周血单个核细胞(peripheral blood mononuclear cell, PBMC),并在10%胎牛血清的T009培养基中培养。该研究由华大(深圳)伦理审查委员会(编号:BGI-IRB18142)批准。
1.2 突变选择和表位预测
从国际癌症基因组协会(ICGC)数据库(http:// icgc.org)中下载中国大肠癌项目(COCA-CN, https:// icgc.org/icgc/cgp/73/371/1001733)的321名结直肠癌患者的体细胞突变数据进行分析。根据分析标准,过滤错义突变,从突变位点中选取体细胞突变表位候选肽和野生型肽。再通过NetMHC-4.0[12]、NetMHCpan-3.0[13]、NetMHCpan-4.0[14]、PSSMHCpan-1.0[15]、PickPocket-1.0[16]和SMM[17]分析候选抗原肽与HLA-A*1101分子之间亲和力。检测结果用IC50作为预测的平衡结合常数(50%抑制浓度,nmol/L),其中IC50<50 nmol/L,表示强亲和力,IC50介于50~500 nmol/L之间,则表示弱亲和力[18]。此外,本研究还使用华大自主开发的Epitope Presentation Integrated Prediction(EPIP)软件[19,20],预测候选抗原肽的呈递,预测结果以最高呈递概率的分数来表示。选择所预测具有强亲和力和高呈递效率的突变肽,作为候选新抗原。
1.3 制备成熟树突状细胞(mature dendritic cell, mDC)
预测的候选多肽与阳性对照肽KRAS G12V8-16(VVGAVGVGK),均由南京金斯瑞生物科技有限公司合成,纯度大于98%。根据文献已报道方法制备mDCs[21],即从健康自愿者分离PBMC,利用CD14 MicroBeads (Miltenyi Biotec公司,德国),富集CD14+细胞,在添加2%人血清白蛋白(WAKO公司,日本)、粒细胞-巨噬细胞集落刺激因子(R&D Systems公司,美国)和IL-4(PeproTech公司,美国)的CellGenixTM DC培养基(CellGenix)中培养5 d。第6 d,添加TNF-α(R&D)、IL-6(PeproTech公司,美国)、IL-1β (R&D Systems公司,美国)、前列腺素E2 (Sigma- Aldrich公司,美国)和Poly(I:C)(InvivoGen公司,美国),继续培养 2 d,即为mDCs。收获后,将mDCs与多肽(1 μg/mL)在37℃无血清的RPMI-1640培养基中孵育4 h,作为抗原呈递细胞。
1.4 诱导新抗原特异性CTLs
从健康自愿者分离PBMC后,用CD8 MicroBeads (Miltenyi Biotec公司,德国)富集CD8+T细胞,再用负载肽的mDC刺激CD8+T细胞(CD8+T:mDC,4∶1),使用上海倍谙基公司的HIPP™-T009淋巴无血清培养基+10%胎牛血清(T009+10%胎牛血清)+ IL21(PeproTech公司,美国)在37℃、5%CO2培养箱中共培养12 d。在共培养的第3 d,向培养基中添加IL-2 (PeproTech公司,美国)、IL-7 (PeproTech公司,美国)和IL-15 (PeproTech公司,美国),每2~3 d更换一次培养基。培养12 d后,用相同负载多肽的mDC再次刺激CD8+T细胞,之后再共培养12 d,最后获得新抗原特异性CTLs。
1.5 酶联免疫斑点法(enzyme-linked immunospot assay, ELISPOT)测定IFN-γ
使用Human IFN-gamma ELISpot PLUS kit (Mabtech公司,瑞典)试剂盒检测IFN-γ的分泌。将2轮扩增后的CTL细胞更换至T009+10%胎牛血清培养基中,静息培养24 h后,按比例与T2细胞(已负载10 μg/mL多肽/未负载多肽)混合,在预处理的ELISPOT板(Mabtech公司,瑞典)内共培养24 h,每个样品设置重复。按照操作说明检测IFN-γ的分泌,用ELISPOT读取器BioReader 4000 (BioSys公司,德国)对斑点成像并计数。根据特异性斑点的数量大于10且高于阴性对照组斑点数2倍以上,判断为阳性反应[22]。
1.6 荧光激活细胞分选(fluorescence activated cell sorting, FACS)
收集细胞,用含有2%胎牛血清的PBS洗涤(FACS缓冲液)后重悬。利用文献报道的方法制备获得APC偶联的pMHC四聚体(pMHC-tetramer- Allophycocyanin (APC),简称pMHC-tetramer-APC)[23]以及购买的商品化PE荧光的抗CD8抗体(BD)、PE荧光的抗mouse-TCRβ抗体(BD),对细胞进行染色,4℃避光45 min后,再用FACS缓冲液洗涤5次。使用FACS AriaⅡ(BD Biosciences公司,美国)流式仪对细胞进行分析,并对CD8+/pMHC四聚体+双阳性T细胞进行分选,进行单细胞RNA测序。使用FlowJo软件分析流式数据。
1.7 通过单细胞RNA测序分析新抗原特异性T细胞受体库(TCR库)
根据Chromium™单细胞V(D)J试剂盒(10x Genomics公司,美国)操作说明,将分选出的CD8+/ pMHC四聚体+双阳性T细胞,通过Chromium™单细胞控制器捕获并分配到乳液凝胶珠(gel bead in emulsion, GEM)中。单细胞和凝胶珠在GEM中裂解后,进行逆转录聚合酶链反应(RT-PCR)扩增,并将barcode标记在mRNA的Poly-A尾上。GEM断裂后,将带barcode的cDNA分子混合到一起,通过PCR扩增富集TCR cDNA的全长V(D)J片段,并进行Illumina®测序建库。通过Cell Ranger™分析方法,分析TCR库及配对的TCR。
1.8 构建TCR-T细胞
将测序获得的TCR的α链、β链的可变区分别与鼠源的恒定区融合,再用T2A序列将α链和β链进行连接,构建进慢病毒表达载体pLVX (CMV- EF1a-ZsGreen-P2A-Bsd)中。如前所述,通过HEK-293细胞,包装制备用于T细胞感染的慢病毒颗粒[24]。慢病毒感染已激活2 d的CD8+T细胞,之后在含10%胎牛血清、IL-2、IL-7和IL-15的T009培养基培养5~7 d,即完成TCR-T制备。
2 结果与分析
2.1 突变候选新抗原的预测
从ICGC数据库下载COCA-CN项目中截止至2018年8月29日的321例结直肠癌患者体细胞突变数据,分析获得3500个单核苷酸变异(single nucleotide variants, SNVs),以及191个InDels突变,共3691个突变位点。每个突变选取突变位点及上游9个氨基酸和下游9个氨基酸,组成共19个氨基酸的突变表位。分析这些突变表位中9~10-mer含突变位点的候选新抗原与对照抗原,共获得60,169个SNVs候选新抗原和6891个InDels候选新抗原。
利用NetMHC-4.0、NetMHCpan-3.0、NetMHCpan- 4.0、PSSMHCpan-1.0、PickPocket-1.0和SMM等软件,预测候选新抗原与HLA-A*1101分子的结合亲和力。一共筛选出56个候选新抗原,它们满足至少3种软件预测显示亲和力IC50均<50 nmol/L,选取3个软件中最小的亲和力预测值作为肽-MHC复合物的亲和力预测值(表1)。此外,还使用了自主研发的HLA-I呈递表位的预测软件算法EPIP,评估预测了强结合亲和力的56个突变表位的呈递概率。在这56个突变表位中,有25个EPIP评分值超过0.90,表明其呈递概率高,因此最终选择这25个突变表位(来源于21个基因)作为候选新抗原(表1)。
2.2 检测CTL体外分泌IFN-γ验证新抗原的免疫原性
通常可以从肿瘤浸润淋巴细胞(TILs)、癌症患者的外周记忆淋巴细胞、健康供体的外周幼稚淋巴细胞中检测到靶向肿瘤新抗原的T细胞[11,25]。相比患者,健康供体来源的PBMC较容易获得,用于鉴定预测新抗原的免疫原性。因此,本研究选择抽取健康人志愿者的外周血,来验证预测的新抗原能否刺激他们的CTL分泌IFN-γ,确认其是否具有免疫原性。用负载候选新抗原的mDC与未负载多肽的mDC,分别与体外PBMC分离的HLA-A*1101分型的CD8+T共培养两轮,获得新抗原特异性的CTL。将静息后的CTL分别与负载对应新抗原的T2细胞共孵育,检测INF-γ的分泌。
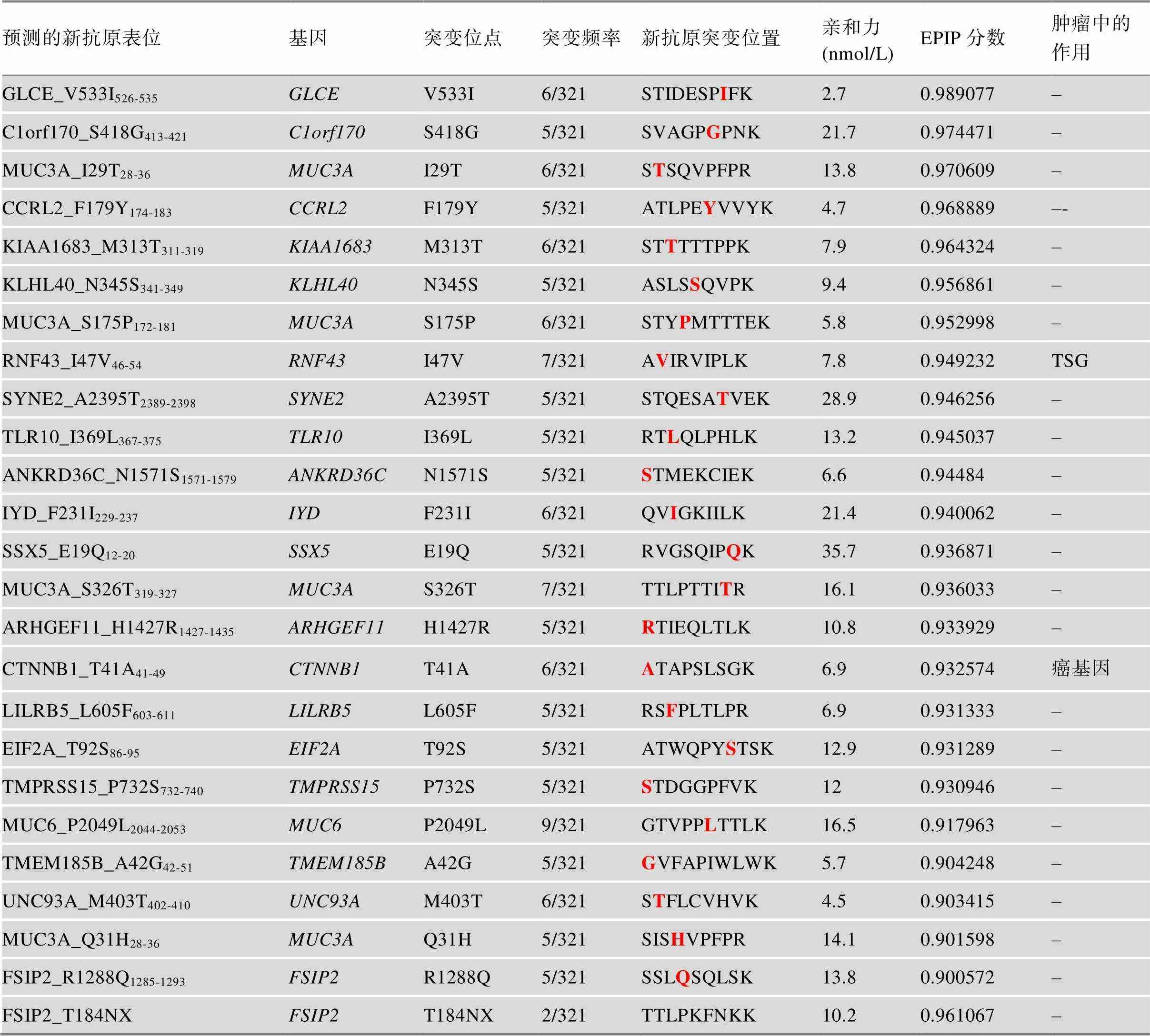
表1 预测的结直肠癌候选抗原肽列表
红色粗体:突变位点;TSG:肿瘤抑制基因。
结果发现,T2细胞负载C1orf170_S418G413-421、KIAA1683_M313T311-319、SSX5_E19Q12-20、TMEM185B_ A42G42-51和UNC93A_M403T402-410等候选抗原肽,与阳性对照抗原肽KRAS_G12V8-16一样,能显著特异性刺激CTL产生IFN-γ,产生斑点数比阴性对照组多10倍(图1,A和B)。另外,T2细胞负载GLCE_V533I526-535、CCRL2_F179Y174-183、ANKRD36C_ N1571S1571-1579、MUC3A_S326T319-327、ARHGEF11_ H1427R1427-1435和MUC6_P2049L2044-2053等抗原肽,也能特异性刺激CTL产生低水平的IFN-γ,比阴性对照多2倍,其余多肽刺激的斑点数与阴性没有明显差异(图1)。总体而言,在25个预测的候选新抗原肽中,有11个具有免疫原性,能特异性刺激CTL分泌IFN-γ,阳性率为44% (11/25)。
2.3 检测并富集新抗原肽特异性CTL
为确定2轮特异性扩增之后新抗原特异性CTL的比例,选择C1orf170_S418G413-421、KIAA1683_ M313T311-319、SSX5_E19Q12-20、TMEM185B_A42G42-51和UNC93A_M403T402-410这5个能显著特异性刺激CTL分泌IFN-γ的候选抗原肽,以及阳性对照抗原肽KRAS_G12V8-16(图2),制备pMHC-tetramer-APC。同时用pMHC-tetramer-APC和CD8-PE荧光抗体对特异性刺激的CTL进行染色,发现抗原肽特异性的CTL的频率范围为0.22%~7.08% (图2A)。根据诱导T细胞分泌IFN-γ的能力,最终选择KRAS_G12V8-16和C1orf170_S418G413-421特异的CTL (图2B)分选后进行单细胞测序,构建TCR库。
2.4 单细胞RNA测序获得抗原肽特异性TCR库
为研究识别HLA-A*1101呈递候选抗原肽C1orf170_S418G413-421的TCR库,从17,800细胞中分选到9243个阳性T细胞,分别得到14,042个α链和9346个β链氨基酸序列,其中产生了8826对TCR (α链与β链配对),多样性为298。序列中,基因(55.38%)和基因(37.35%)占主要比例(图3A);序列中,基因占99.88% (图3B)。CDR3α氨基酸长度分布主要在12-mer (55.64%)和15-mer (39.09%) (图3C)。而序列中,基因占87.47% (图3D),序列中,占99.91% (图3E)。CDR3β氨基酸长度主要分布为15-mer (88.04%) (图3F)。12-mer和15-mer CDR3α高度保守氨基酸基序分别是CAASGGAQKLVF(图3G)和CAGLLYNSGNTPLVF (图3H),与和相一致,而CDR3β高度保守基序为CASSRDRGSNQPQHF (图3I),与相一致。
最终获得2个在TCR库中占主要比例的克隆,53.2% (4694/8826)T细胞(Clonotype1) TCRα为和,TCRβ为;31.6% (2786/8826) T细胞(Clonotype2)表达的TCRα为,表达的TCRβ为(表2)。
为获得HLA-A*1101呈递阳性抗原肽KRAS_ G12V8-16的TCR库,从21,000个细胞中分选到12,530个阳性T细胞,最终得到11,137个α链和13,126个β链氨基酸序列,其中产生了10,559对TCR (α链与β链配对),多样性为1610。序列中,基因占81.69% (图4A);序列中,基因占99.69% (图4B)。CDR3α氨基酸长度主要分布在10-mer (81.88%) (图4C)。而序列中,基因占83.35% (图4D),序列中,基因占99.81% (图4E)。CDR3β氨基酸长度主要分布分别在14-mer (86.4%) (图4F)。并且10-mer CD3α高度保守氨基酸基序是CASNDYKLSF (图4G),与的序列一致;14-mer CDR3β高度保守氨基酸基序是CASSLDGVSYEQYF (图4H),与的序列一致。
最终获得2个在TCR库中占很大比例的克隆,80.5% (8499/10559) T细胞(Clonotype1) TCRα为,TCRβ为;14.3% (1515/10559)T细胞(Clonotype2) TCRβ为,但是未检测到表达的TCRα序列(表2)。实验结果表明,占主要丰度的TCR基因序列,能够代表对应TCR库的基因序列组成。
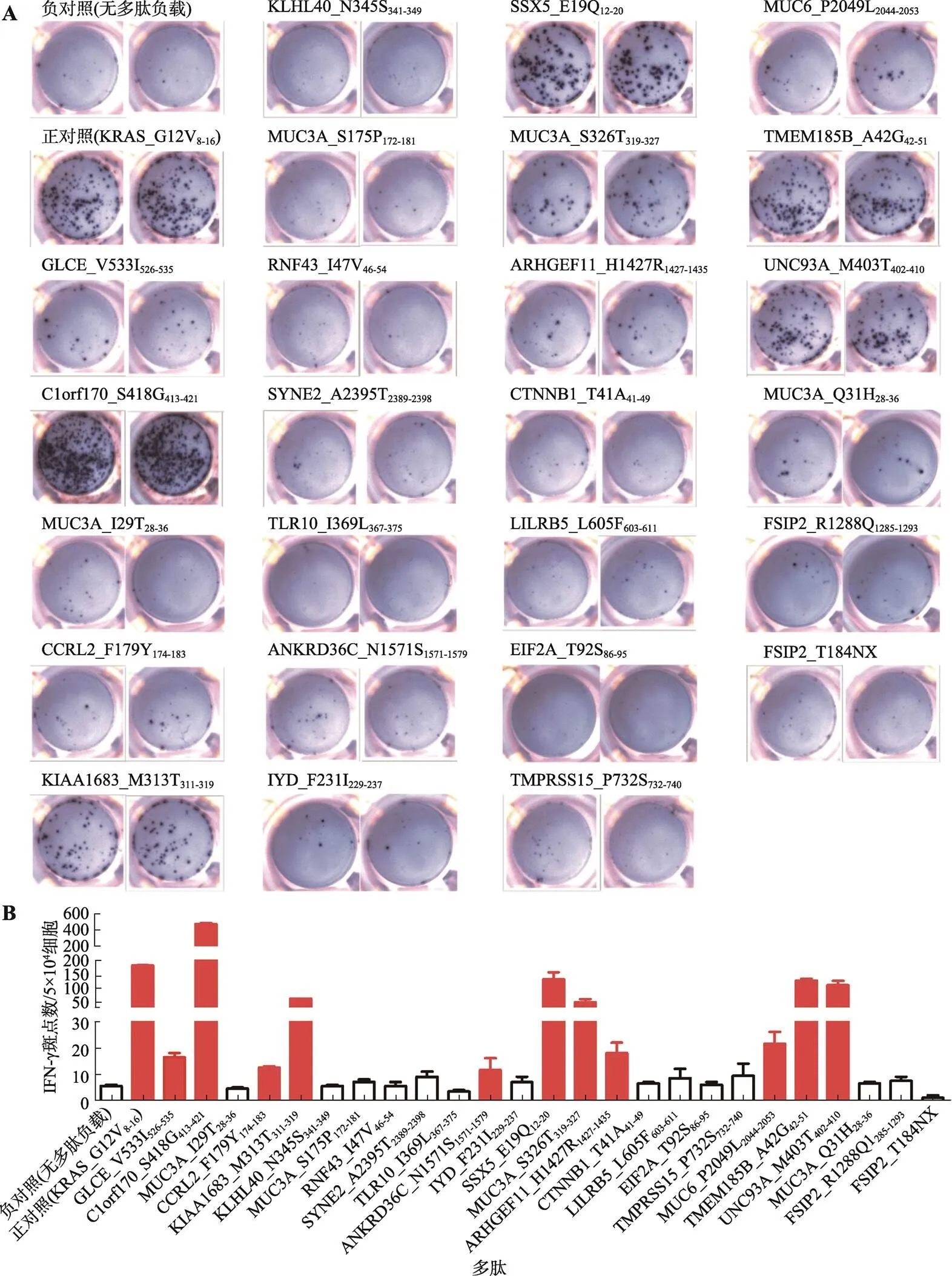
图1 T2细胞负载候选多肽刺激CTL分泌IFN-γ
:T2细胞分别负载不同抗原肽,刺激相应新抗原特异性CTL分泌IFN-γ;B:实验组及对照组产生IFN-γ斑点数的统计。红色柱形代表产生的斑点数比阴性多2倍以上,有显著性差异;白色柱形代表产生的斑点数与阴性无显著性差异。
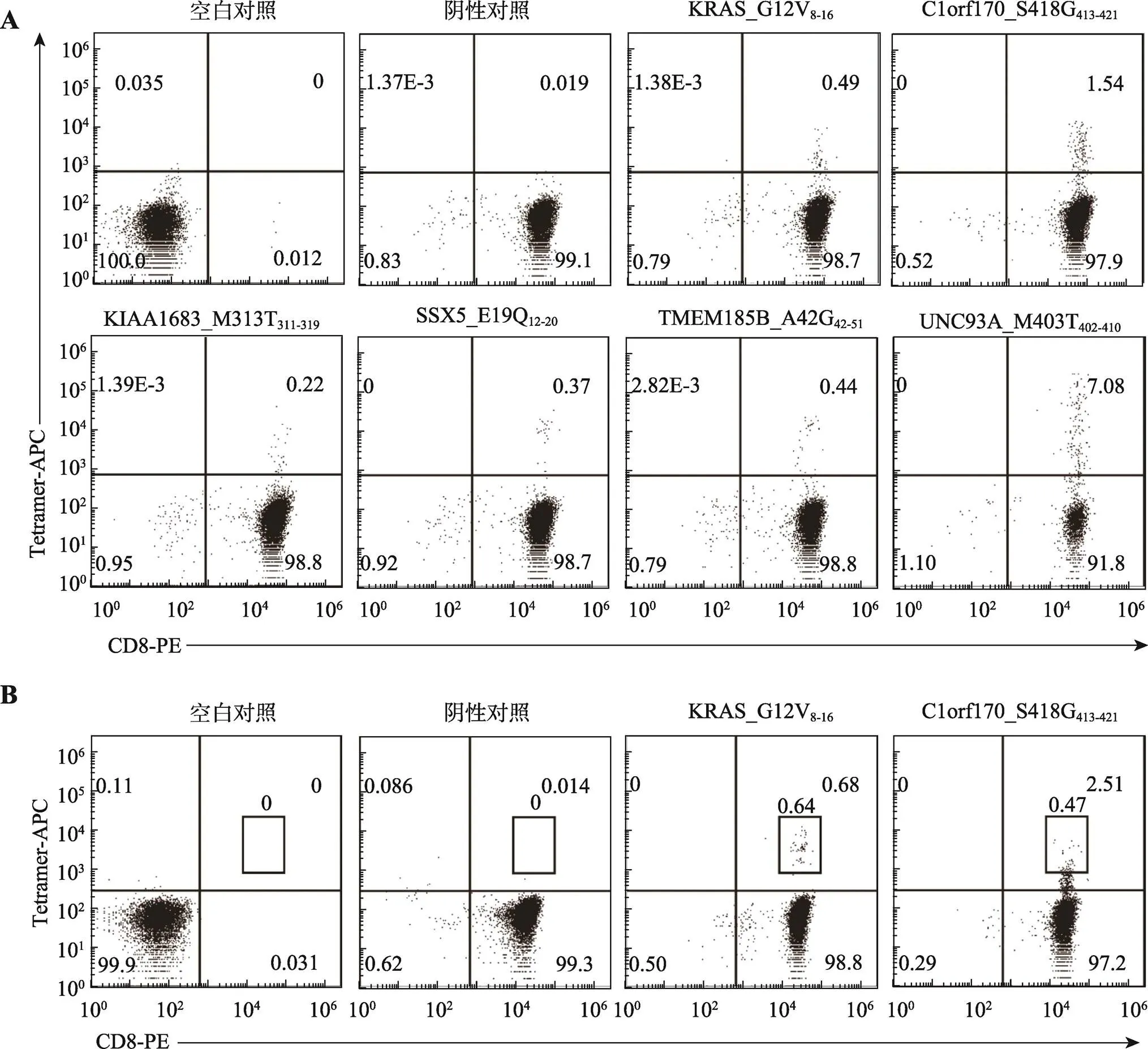
图2 流式检测候选抗原肽特异性CTL的比例
A:扩增2轮后的新抗原特异性CTL阳性率检测;B:分选新抗原特异性的CTL进行单细胞测序建库。
2.5 抗原肽特异性的T细胞克隆功能验证
从TCR库中选择比例最高的2个TCR序列(表2),构建TCR-T并验证功能。流式结果显示:C1orf170_S418G413-421特异性TCR-T阳性率为84.1%,KRAS_G12V8-16特异性TCR-T阳性率为68.8% (图5A)。T2细胞负载抗原肽C1orf170_S418G413-421能特异性激活对应的TCR-T细胞,产生明显的IFN-γ阳性斑点(图5B,C1orf170组左上),而刺激未过表达TCR的CD8+T组仅产生少量背景斑点(图5B C1orf170组左下),两者有显著性差异,证实此TCR有功能;并且,对比T2细胞负载野生型C1orf170抗原肽(图5B,C1orf170组中)或无关多肽(图5B,C1orf170组右)刺激TCR-T和CD8+T对照组,其斑点数明显增多(图5,B和C),说明TCR的特异性。T2细胞负载阳性对照抗原肽KRAS_G12V8-16,也能特异性刺激TCR-T细胞产生IFN-γ阳性斑点,与阴性对照组有明显差异(图5,B和C)。因此,本研究通过建立的结直肠癌新抗原特异性T细胞克隆筛选流程,最终获得2个预测候选抗原肽特异性的TCR克隆,并且具有明显的细胞免疫功能。相关数据已上传至CNGB数据库CNSA (https://db.cngb.org/cnsa/),登录号为CNP0000518。
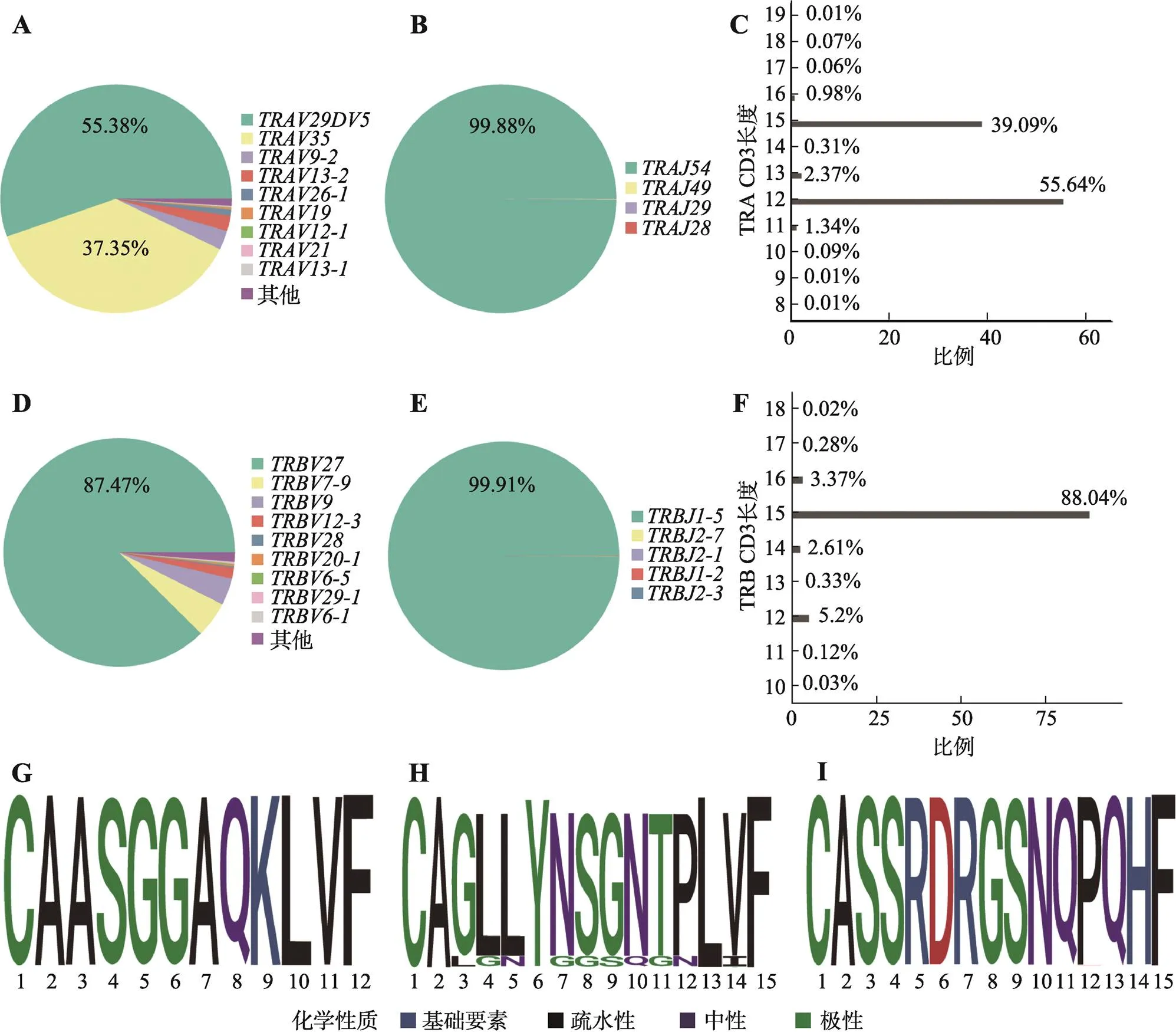
图3 识别HLA-A*1101限制性新抗原C1orf170_S418G413-421的TCR库特征分析
A:基因序列的组成;B:基因序列的组成;C:TRA CDR3氨基酸长度分布;D:基因序列的组成;E:基因序列的组成;F:TRB CDR3氨基酸长度分布;G:12-mer CDR3α高度保守氨基酸基序;H:15-mer CDR3α高度保守氨基酸基序;I:CDR3β高度保守氨基酸基序。
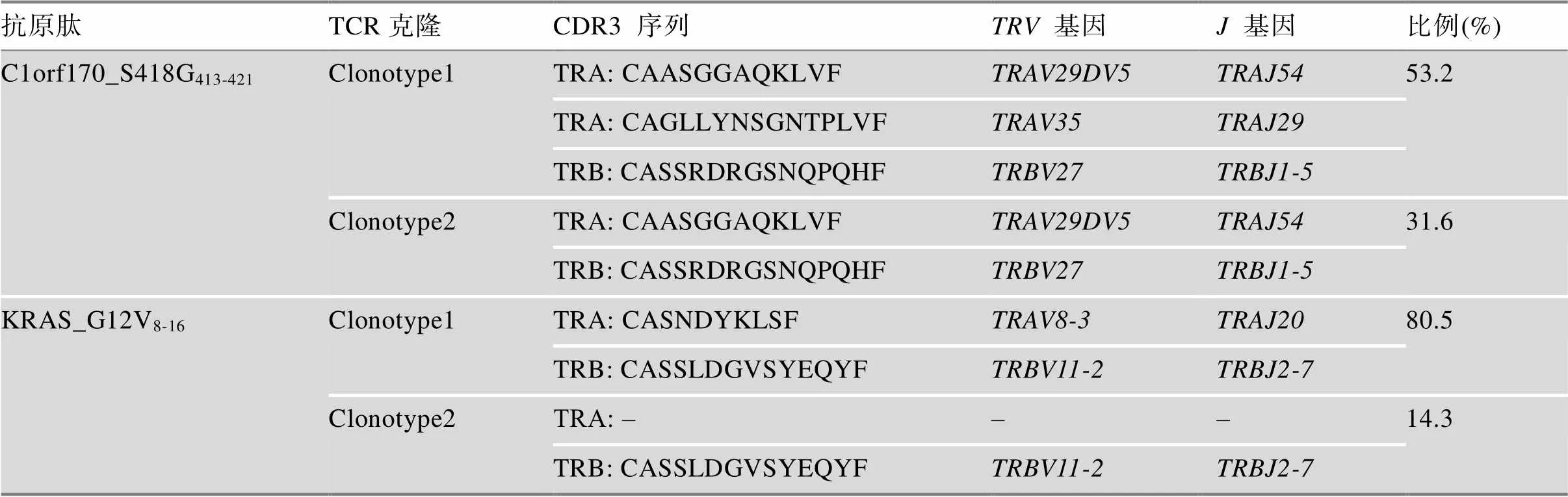
表2 两个不同TCR库中丰度前两位的TCR α和β链配对信息
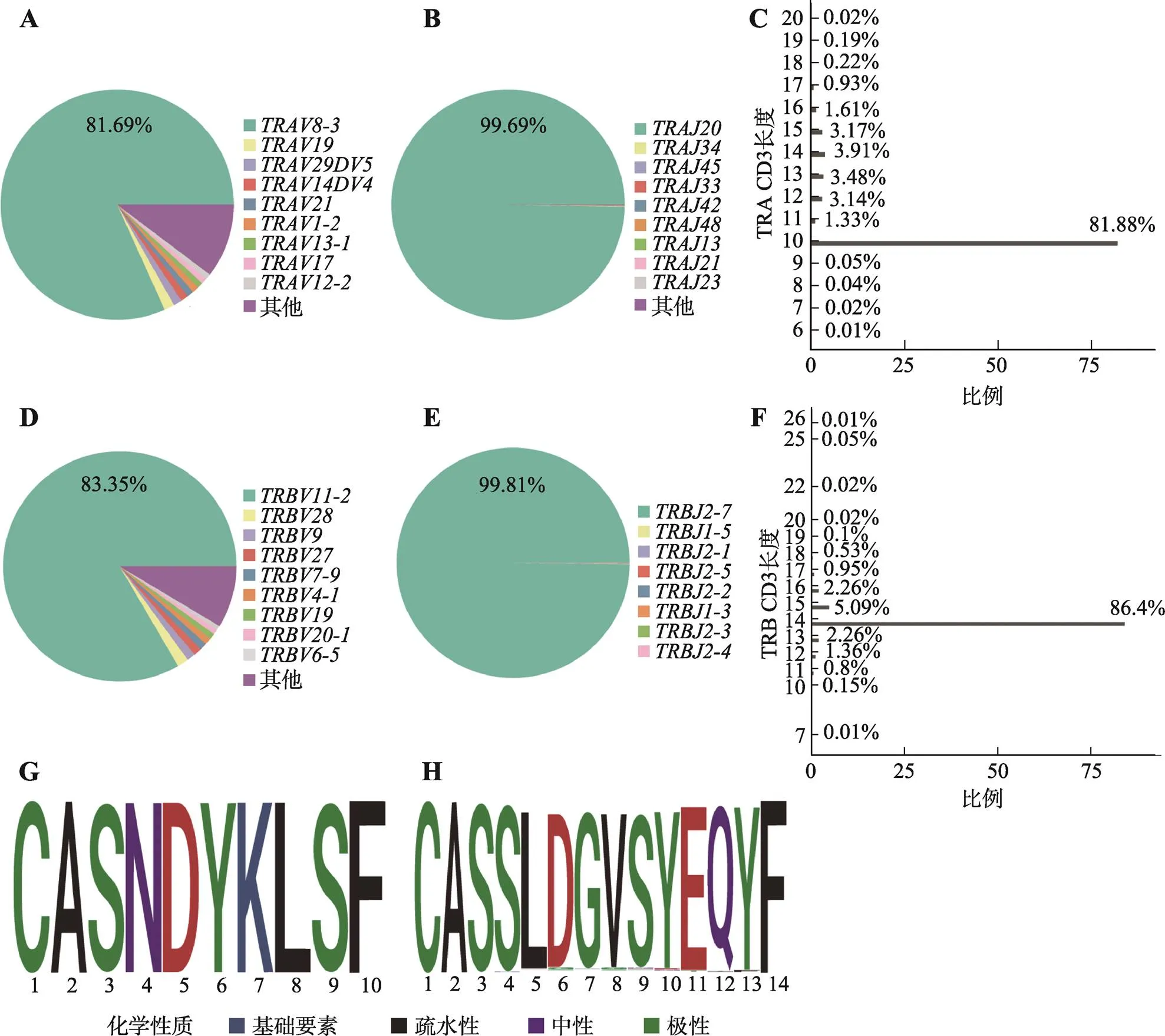
图4 识别HLA-A*1101限制性新抗原KRAS_G12V8-16的TCR库特征分析
A:基因序列的组成;B:基因序列的组成;C:TRA CDR3氨基酸长度分布;D:基因序列的组成;E:基因序列的组成;F:TRB CDR3氨基酸长度分布;G:10-mer CDR3α高度保守氨基酸基序;H:14-mer CDR3β高度保守氨基酸基序。
3 讨论
肿瘤在发生和发展过程中常会获得大量的体细胞突变,突变基因被翻译成蛋白质后,经蛋白酶体水解被MHC分子呈递在细胞表面,产生新的抗原[10]。肿瘤突变相关的新抗原在正常组织中并不存在,是肿瘤特有的生物标记物[10]。T细胞识别新抗原并不受胸腺选择和中枢耐受性的影响,所以人体内可能会存在高亲和力的新抗原特异性T细胞[7,10]。因此,利用全外显子、RNA测序并结合生物信息学流程,可以从单核苷酸分辨率水平揭示肿瘤特异性的改变,并预测用于癌症免疫治疗的新抗原[26]。在确定错义突变和基因表达水平后,使用各种算法可以评估、预测新抗原多肽与MHC结合的亲和力和呈递能力[20]。然而研究发现绝大多数预测的新抗原仅有少数可以引发T细胞反应[27]。Chizu等[8]通过NetMHC4.0算法预测了26个突变表位与HLA-A*2402具有较强的结合能力,但利用5种癌症患者来源的PBMC仅鉴定出1种多肽具有免疫原性。因此,我们认为当前的算法并不理想,可以同时使用多种算法来提高多肽结合亲和力预测的准确性。
本研究使用了多种多肽-MHC结合亲和力预测算法,包括NetMHC-4.0、NetMHCpan-3.0、NetMHCpan-4.0、PSSMHCpan-1.0、PickPocket-1.0和SMM,以及自主开发的预测表位呈递的EPIP算法来评估筛选多肽信息,最终获得25个候选多肽。它们同时满足了以下条件:SNVs出现频率为321名患者中超过5人以上(SNVs>5/321),InDels出现频率为321个患者中超过2个以上(InDels>2/321),至少3种软件预测的IC50值小于50 nmol/L并且EPIP分值≥0.90 (表1),来评估其呈递和诱导细胞毒性T细胞的特征。根据ELISPOT实验结果,25个候选多肽中有11个(44%)新抗原可诱导特异性CTL分泌IFN-γ (图1)。

图5 所构建C1orf170_S418G413-421和KRAS_G12V8-16特异性TCR-T的免疫应答功能验证
A:流式方法分析实验组TCR-T与对照组GFP-T的阳性率;B:ELISPOT方法检测实验组TCR-T与对照组CD8+TIFN-γ的分泌;C:实验组TCR-T与对照组CD8+TIFN-γ分泌差异的统计。*:表示有统计学差异;:表示无统计学差异。
预测新抗原的免疫原性通常利用TIL的应答来进行检测[10,11]。虽然外周血中新抗原反应性T细胞出现的频率通常低于肿瘤样品,来源于癌症患者的循环CD8+记忆性T细胞或健康志愿者的循环CD8+初始T细胞的新抗原特异性T细胞,可以通过与负载新抗原的同源DC细胞在体外共培养后富集,利用常规实验方法即可检测它们识别新抗原的应答能力[11,25,28]。因此,本研究采用较易获得的健康供体的循环CD8+T细胞与负载新抗原的同源DC共培养两轮后,来验证所预测新抗原的免疫原性。除阳性表位KRAS_G12V8-16外,发现25个预测的抗原中有11个(44%)负载T2细胞后可诱导共培养的CTL分泌IFN-γ (图1),并且产生IFN-γ斑点数超过阴性对照的10倍以上的特异性CTL频率在0.22%~7.08%之间(图2A)。其余14个候选新抗原(14/25)并未发现具有免疫原性,推测是由于一般淋巴结10E5~10E6个T细胞中才大约只有1个T细胞为新抗原特异性T细胞,而健康供体循环CD8+记忆T细胞并不会产生新抗原反应性T细胞。因此,在抗原致敏(antigen priming)前富集健康志愿者的循环CD8+初始T细胞,可以增加特定T细胞遇到呈递相关新抗原的DC的可能性[25,29]。此外,由于依赖供体的T细胞群存在个体间差异和人类TCR库的足够多样性,需要筛选多个供体以鉴定候选新抗原的免疫原性[25]。因此,其余免疫原性阴性的新抗原多肽,可能会从其他健康志愿者筛选获得的阳性的CTL。
在新抗原特异性TCR-T的功能验证部分,本研究通过对比T2负载新抗原多肽、野生型多肽以及无关多肽与TCR-T共孵育所产生的ELISPOT斑点,来说明所筛选到的TCR的特异性。实验发现C1orf及KRAS两组T2负载野生型多肽或无关多肽组也能刺激TCR-T或CD8+T细胞产生少量斑点,可能是由于部分TCR-T或CD8+T细胞为旁观者激活的T细胞(bystander activated T cells),处于非特异性的激活状态,可分泌IFN-γ细胞因子产生非特异性背景信号[30~32]。此外,通过前期实验也发现不同的健康志愿者,甚至同一个健康志愿者不同时间获取的T细胞,所做ELISPOT背景信号都有差异,分析是由于不同个体或者同一个体不同时间的免疫状态的差异,导致背景中旁临激活的T细胞含量不同产生的。这些信号均为背景信号,并不影响判断TCR-T有效性的整体趋势。此外,即使在实验过程中没有筛选到突变和野生型抗原组有差别的特异性TCR,从另一角度也证明了此功能性TCR筛选方法的可靠性。
综上所述,本研究获得了结直肠癌中HLA-A* 1101分型C1orf170_S418G413-421的TCR-T,为结直肠癌过继TCR-T细胞疗法提供了可能,同时证实从体细胞突变数据库中分析获得高频且有免疫原性新抗原的方法可行性,可将获得的新抗原开发为多肽、DC等疫苗以及TCR-like抗体的结直肠癌免疫治疗通用靶点。同时,此方法也为其他癌种免疫治疗靶点的选择提供了可靠的科研方案,具有重要的临床意义。
[1] Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries., 2018, 68(6): 394–424.
[2] Magee MS, Kraft CL, Abraham TS, Baybutt TR, Marszalowicz GP, Li P, Waldman SA, Snook AE. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity., 2016, 5(10): e1227897.
[3] Ciardiello D, Vitiello PP, Cardone C, Martini G, Troiani T, Martinelli E, Ciardiello F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy., 2019, 76: 22–32.
[4] Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis., 2011, 19(3): 620–626.
[5] Zhang CC, Wang Z, Yang Z, Wang ML, Li SQ, Li YY, Zhang R, Xiong ZX, Wei ZH, Shen JJ, Luo YL, Zhang QZ, Liu LM, Qin H, Liu W, Wu F, Chen W, Pan F, Zhang XQ, Bie P, Liang HJ, Pecher G, Qian C. Phase I escalating- dose trial of CAR-T therapy targeting CEA+metastatic colorectal cancers., 2017, 25(5): 1248–1258.
[6] Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2., 2010, 18(4): 843–851.
[7] Zhou Z, Lyu XZ, Wu JC, Yang XY, Wu SS, Zhou J, Gu X, Su ZX, Chen SQ. TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection., 2017, 4(4): 170050.
[8] Nonomura C, Otsuka M, Kondou R, Iizuka A, Miyata H, Ashizawa T, Sakura N, Yoshikawa S, Kiyohara Y, Ohshima K, Urakami K, Nagashima T, Ohnami S, Kusuhara M, Mitsuya K, Hayashi N, Nakasu Y, Mochizuki T, Yamaguchi K, Akiyama Y. Identification of a neoantigen epitope in a melanoma patient with good response to anti- PD-1 antibody therapy., 2019, 208: 52–59.
[9] Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, Kriley IR, Rosenberg SA. T-Cell transfer therapy targeting mutant KRAS in cancer., 2016, 375(23): 2255– 2262.
[10] Wirth TC, Kühnel F. Neoantigen targeting-dawn of a new era in cancer immunotherapy?, 2017, 8: 1848.
[11] Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, Gartner JJ, Jia L, Ray S, Ngo LT, Jafferji M, Sachs A, Prickett T, Robbins PF, Rosenberg SA. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients., 2019, 10(1): 449.
[12] Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable predictionof T-cell epitopes using neural networks with novel sequence representations., 2003, 12(5): 1007–1017.
[13] Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets., 2016, 8(1): 33.
[14] Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved Peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data., 2017, 199(9): 3360–3368.
[15] Liu G, Li DL, Li Z, Qiu S, Li WH, Chao CC, Yang NB, Li HD, Cheng Z, Song X, Cheng L, Zhang XQ, Wang J, Yang HM, Ma K, Hou Y, Li B. PSSMHCpan: a novel PSSM-based software for predicting class I peptide-HLA binding affinity., 2017, 6(5): 1–11.
[16] Zhang H, Lund O, Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding., 2009, 25(10): 1293–1299.
[17] Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method., 2005, 6: 132.
[18] Creaney J, Ma S, Sneddon SA, Tourigny MR, Dick IM, Leon JS, Khong A, Fisher SA, Lake RA, Lesterhuis WJ, Nowak AK, Leary S, Watson MW, Robinson BW. Strong spontaneous tumor neoantigen responses induced by a natural human carcinogen., 2015, 4(7): e1011492.
[19] Hu WP, Li YP, Zhang XQ. MHC-I epitope presentation prediction based on transfer learning.41(11): 1041–1049.胡伟澎, 李佑平, 张秀清. 基于迁移学习的MHC-I型抗原表位呈递预测.遗传, 41(11): 1041–1049.
[20] Hu WP, Qiu S, Li YP, Lin XX, Zhang L, Xiang HT, Han X, Chen L, Li S, Li WH, Ren Z, Hou GX, Lin ZL, Lu JL, Liu G, Li B, Lee LJ. EPIP: MHC-I epitope prediction integrating mass spectrometry derived motifs and tissue- specific expression profile., 2019, 567081.
[21] Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, Baumgartner P, Stevenson BJ, Iseli C, Dangaj D, Czerniecki B, Semilietof A, Racle J, Michel A, Xenarios I, Chiang C, Monos DS, Torigian DA, Nisenbaum HL, Michielin O, June CH, Levine BL, Powell DJ Jr, Gfeller D, Mick R, Dafni U, Zoete V, Harari A, Coukos G, Kandalaft LE. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer., 2018, 10(436): eaao5931.
[22] Yamamiya D, Mizukoshi E, Kaji K, Terashima T, Kitahara M, Yamashita T, Arai K, Fushimi K, Honda M, Kaneko S. Immune responses of human T lymphocytes to novel hepatitis B virus-derived peptides., 2018, 13(6): e0198264.
[23] Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange., 2006, 1(3): 1120–1132.
[24] Kim MS, Ma JS, Yun HY, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, Gonzalez J, Schultz PG, Young TS, Kim CH. Redirection of genetically engineered CAR-T cells using bifunctional small molecules., 2015, 137(8): 2832–2835.
[25] Ali M, Foldvari Z, Giannakopoulou E, Böschen ML, Strønen E, Yang W, Toebes M, Schubert B, Kohlbacher O, Schumacher TN, Olweus J. Induction of neoantigen- reactive T cells from healthy donors., 2019, 14(6): 1926–1943.
[26] Lancaster EM, Jablons D, Kratz JR. Applications of next-generation sequencing in eoantigen prediction and cancer vaccine development., 2019, 24(2): 59–66.
[27] The problem with neoantigen prediction., 2017, 35(2): 97.
[28] Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, Li YF, El-Gamil M, Rosenberg SA, Robbins PF. Isolation of neoantigen- specific T cells from tumor and peripheral lymphocytes., 2015, 125(10): 3981–3991.
[29] Lin QY, Liu Z, Luo MJ, Zheng H, Qiao S, Han CL, Deng DQ, Fan Z, Lu YF, Zhang ZH, Luo QM. Visualizing DC morphology and T cell motility to characterize DC-T cell encounters in mouse lymph nodes under mTOR inhibition., 2019, 62(9): 1168–1177.
[30] Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong BH, Koh T, Hillmer AM, Takano A, Lim TKH, Tan EH, Zhai W, Tan DSW, Tan IB, Newell EW. Bystander CD8+T cells are abundant and phenotypically distinct in human tumour infiltrates., 2018, 557(7706): 575–579.
[31] Whiteside SK, Snook JP, Williams MA, Weis JJ. Bystander T Cells: a balancing act of friends and foes., 2018, 39(12): 1021–1035.
[32] Kim TS, Shin EC. The activation of bystander CD8+T cells and their roles in viral infection., 2019, 51(12): 1–9.
A method of screening highly common neoantigens with immunogenicity in colorectal cancer based on public somatic mutation library
Lili Qin1,2, Yijian Li1, Zhaoduan Liang1, Lei Chen1, Wenhui Li1, Chao Chen1,3,4, Yaling Huang1, Le Zhang1,3,4, Songming Liu1,3,4, Si Qiu1,4, Yuping Ge1, Wenting Peng1,3, Xinxin Lin1,4, Xiuqing Zhang1,4, Xuan Dong1, Bo Li1,4
Colorectal cancer (CRC) is a malignant cancer with high incidence and mortality in the world. Immunotherapy targeting neoantigens can induce durable tumor regression in cancer patients, but is almost limited to personalized precision therapy, due to the individual differences of unique neoantigens. With the discovery of many common oncogenic mutations, and such mutation-associated neoantigens could cover more patients, and hence are valuable in clinical field. However, whether the common neoantigens can be identified in CRC is unknown. Combining the somatic mutations data from 321 CRC patients with a filter standard and 7 predicted algorithms, we screened and obtained 25 HLA-A*1101-restricted common neoantigens with a high binding affinity (IC50<50 nmol/L) and presentation score (>0.90). Besides the positive epitope KRAS_G12V8-16, 11 out of 25 common neoantigens specifically inducedpre- stimulated cytotoxic lymphocyte (CTL) to secrete interferon gamma (IFN-γ). Moreover, combining cell-sorting technology and single-cell RNA sequencing, the immune repertoire profiles of C1orf170_S418G413-421and KRAS_G12V8-16-specific CTL were analyzed and validated. Their related T-cell receptor engineered T cell (TCR-T) cells could also recognize the neoantigens and secrete IFN-γ. Hence, we have established a method to screen for common neoantigens with immunogenicity in CRC based on the public somatic mutation library. It can provide essential peptide and TCR information for immunotherapies, such as peptides, dendritic cells (DC) vaccines, TCR-like antibodies, TCR-T, etc., for the CRC and other cancers, which has practical application value in the clinics.
colorectal cancer; common; neoantigen; presentation
2020-04-10;
2020-05-15
国家自然科学基金项目(编号:81702826)和深圳市科技创新委员会项目(编号:JCYJ20170303151334808, JCYJ20170817150015170)资助[Supported by the National Natural Science Foundation of China (No. 81702826), and Science, Technology and Innovation Commission of Shenzhen Municipality (Nos. JCYJ20170303151334808, JCYJ20170817150015170)]
秦丽丽, 在读硕士研究生,专业方向:病理学和病理生理学。E-mail: veritasdoct168@163.com李毅坚,硕士,助理研究员,研究方向:肿瘤免疫治疗。E-mail: liyijian@genomics.cn秦丽丽和李毅坚为并列第一作者。
董旋,博士,副研究员,研究方向:肿瘤免疫治疗。E-mail: dongxuan@genomics.cn李波,硕士,研究员,研究方向:基因组学及免疫治疗。E-mail: libo@genomics.cn
10.16288/j.yczz.20-032
2020/5/20 16:44:00
URI: http://kns.cnki.net/kcms/detail/11.1913.r.20200520.0831.001.html
(责任编委: 周钢桥)
猜你喜欢
昆明医科大学学报(2021年1期)2021-02-07
现代园艺(2017年13期)2018-01-19
上海农业学报(2017年4期)2017-04-10
现代检验医学杂志(2016年3期)2016-11-15
特产研究(2016年3期)2016-04-12
中国学术期刊文摘(2016年1期)2016-02-13
医学研究杂志(2015年6期)2015-07-01
药学与临床研究(2015年4期)2015-06-05
科学中国人(2015年16期)2015-02-28
癌变·畸变·突变(2015年3期)2015-02-27
