
组蛋白去乙酰化酶在调节心肌肥大过程中的作用机制
2020-06-29 01:20任恋吴秀山李永青
遗传 2020年6期
任恋,吴秀山,李永青
综 述
组蛋白去乙酰化酶在调节心肌肥大过程中的作用机制
任恋,吴秀山,李永青
湖南师范大学省部共建淡水鱼类发育生物学国家重点实验室,教育部重点实验室,生命科学学院心脏发育研究中心,长沙 410081
心肌肥大(cardiac hypertrophy)是由外周组织对血流动力学需求增加而发生的一种代偿性反应。在心肌肥大过程中,不同时期的不同类型的基因表达受到生理和病理信号的多级转录调控。组蛋白乙酰化作为最广泛的翻译后修饰方式,受相互拮抗的组蛋白乙酰化酶(histone acetyltransferases, HAT)和组蛋白去乙酰化酶(histone deacetylases,HDACs)的精细控制。近年来研究表明,HDACs作为一类抑制转录过程并含有高度保守的脱乙酰酶结构域家族酶,通过多种作用途径调控心肌肥大过程中的基因表达。本文主要综述了组蛋白去乙酰化酶调节心肌肥大过程的相关研究进展,通过阐明不同种类HDACs在心肌肥大中的作用和分子机制,为不同类型心肌肥大和心衰的发病治疗提供新的思路,为新药设计提供分子靶点。
心肌肥大;转录调控;翻译后修饰;组蛋白去乙酰化酶;分子机制
心脏组织对生理和病理刺激的持续反应会导致心脏细胞尺寸增大,被称为心肌肥大(cardiac hypertrophy),是由外周组织对血流动力学需求增加而发生的一种代偿性反应。早期心肌肥大会出现心肌细胞的细胞负荷增加和胎儿基因的重编程等表征。心肌肥大初期可通过增厚心脏壁以应对高血压、心脏损伤、瓣膜疾病和心肌梗塞引起的压力增加,具有一定生理性的代偿意义,但持续的肥大会使心脏功能下降进入病理状态[1],从而导致心肌病、纤维化和心力衰竭等病理表型。
在心肌肥大过程中,不同时期和不同类型的基因表达受到生理和病理信号的多级转录调控。组蛋白乙酰化作为最广泛的翻译后修饰方式,受相互拮抗的组蛋白乙酰化酶(histone acetyltransferase, HAT)和组蛋白去乙酰化酶(histone deacetylases, HDACs)的精细控制,其状态的改变将导致染色体结构发生变化从而影响相关基因的转录。目前研究表明,HDACs既可以通过紧缩染色质而抑制基因的转录,也可以通过调控转录因子活性或酶活性来调控基因的转录[2]。本文介绍了HDACs的分子结构特点,着重对不同种类HDACs在心肌肥大中的作用和分子机制进行综述,并概述了HDACs抑制剂的抗心肌肥大作用,以期为不同类型心肌肥大和心衰的发病治疗提供新的思路,为新药设计提供分子靶点。
1 HDACs的分子结构特点
HDACs家族是一类抑制转录过程并含有高度保守的脱乙酰酶结构域家族酶总称。哺乳动物HDACs家族酶的18个成员被分为I类、IIa类、IIb类、III类和IV类,具有不同的结构、酶功能、亚细胞定位和表达模式以及生物学作用[3~7]。I、II和IV类HDACs是锌(Zn2+)依赖性酶,彼此之间呈现序列相似性,但与III类HDACs没有同源性,后者需要NAD+进行脱乙酰基作用。迄今为止,其他3类HDACs都有成员被发现参与心肌肥大的调节,只有IV类HDAC即HDAC11尚无相关报道。相关内容总结见表1和图1。
2 I类HDACs在心肌肥大中的作用
HDAC2敲除小鼠(Mus musculus)或转基因小鼠以及心肌细胞中的研究结果都已表明,I类HDAC中的 HDAC2对心脏肥大具有正调控作用[13~16]。HDAC2敲除小鼠出生后24 h内死于严重心脏畸形,心肌细胞由于过度增殖导致右心室腔闭塞,并出现心动过缓的现象[13]。HDAC2在调节心肌肥大基因转录过程中起着非常重要的作用[17],相关机制主要集中在两方面:一方面是HDAC2直接作用于抗肥大调节因子,通过下调抗肥大调节因子表达而促进心肌肥大。活化的HDAC2被募集到驱动抗肥大调节因子(如INPP5F或KLF4)的启动子上,通过对抗肥大调节因子启动子处的组蛋白去乙酰化,下调抗肥大调节因子的表达,继而导致被其阻遏的胎儿基因程序的再激活,引起心肌肥大现象[14]。其中HDAC2下调抗肥大调节因子INPP5F的表达后,通过Pdk- Akt-Gsk3β信号通路减轻了Gsk3β对elF2b、c-Myc、GATA4和β-catenin等促肥大因子的抑制而引起心肌肥大[15]。HOPx/HDAC2/HSP70信号下调抗肥大转录因子KLF4的表达,下调的KLF4转录因子能提高肥大靶标基因心房利钠肽前体表达水平从而引起心肌肥大[16]。HDAC2抗心肌肥大的另一个作用机制是可直接作用于转录因子,通过调节转录因子的活性来调控心肌细胞增殖和肥大靶基因的表达。在心肌细胞中,转录因子SRF驱动肥大靶基因表达,SRF的活性和组蛋白翻译后p300乙酰修饰化存在强烈的相关性[51]。SRF的辅因子Myocardin募集p300,诱导组蛋白3乙酰化增强SRF转录因子的活性,促进肥大靶基因表达[52]。心肌细胞中特异表达的Hopx蛋白可以通过招募HDAC2形成转录抑制复合物抑制心肌细胞中SRF转录因子活性来抑制肥大靶基因的表达[18]。另外,Hopx还可增加HDAC2-GATA4复合物的稳定性,导致转录因子GATA4去乙酰化而抑制该转录因子与DNA结合及转录激活的能力,抑制心肌细胞增殖[19]。雷帕霉素(mammalian target of rapamycin, mTOR)激酶是包括生理性和病理性心脏肥大在内的多种状况下细胞生长的核心调节因子。药理和遗传方法的研究证明,抑制I类HDAC通过抑制mTOR活性而抑制病理性心脏肥大,依赖于结节性硬化复合物2 (tuberous sclerosis complex 2, TSC2)的mTOR抑制是I类HDAC抑制剂抑制病理性心肌肥大的关键靶点。HDAC1、2、3对mTOR和心肌细胞肥大生长的调节作用存在功能冗余[8]。

表1 HDACs亚族分子结构特点

图1 HDACs亚族分子在心肌肥大中的作用靶标和功能
Fig. 1 Target and function of HDACs subfamily molecules in myocardial hypertrophy
A:I和II类HDACs在心肌肥大中的作用靶标和功能;B:III类HDACs在心肌肥大中的作用靶标和功能。
HDAC1敲除小鼠胚胎由于严重的增殖缺陷和发育延迟在E10.5之前就会死亡,HDAC1的缺失会诱导HDAC2和HDAC3的表达,但并不能弥补HDAC1的作用[9]。在小鼠心脏中同时特异性敲除HDAC1和HDAC2等位基因会导致胚胎致死,并伴有心脏心律失常、扩张型心肌病、骨骼肌特定收缩蛋白和心脏中钙通道相关基因的表达上调,说明HDAC1与HDAC2在心脏功能上可能存在冗余,单独失活HDAC1或HDAC2 并不能阻断异丙肾上腺素(isoprenaline, ISO)或者主动脉缩窄术(transverse aortic constriction, TAC)引起的肥大,并且同时出现在sin3、NuRD、CoREST和PRC2等抑制复合物中[10]。在高同型半胱氨酸血症患者中,形成HDAC1和肌细胞增强因子2C (myocyte enhancer factor-2C, MEF2C)阻遏复合物,使心肌肥大重要转录因子MEF2C失活,导致心肌细胞中的抗肥大miR-133a的表达下降而诱导心肌肥大[11]。HDAC1还可与HDAC5形成复合物。HDAC5-HDAC1复合体使转录因子Nkx2.5去乙酰化;而去乙酰化的Nkx2.5募集p300结合到Ncx1启动子处上调Ncx1的表达,促进心肌细胞中的钠钙交换引起心肌肥大[12]。
与其他I类HDACs相反,HDAC3对心肌肥大可能主要起抑制作用。HDAC3是唯一一个在体外和体内使MEF2D去乙酰基的I类HDAC;它通过与MEF2的MADS结构域、以及乙酰基转移酶p300以及p300/CBP结合因子(p300/CBP-associated factor, PCAF)直接结合,使MEF2D和PCAF都去乙酰化,从而抑制MEF2依赖性转录和肌生成[21]。在ISO诱导的肥大心肌中,心肌肥大信号的重要激酶p38 MAPK是受乙酰化激活而与其磷酸化状态无关。p300和PCAF使p38的K53和K152发生赖氨酸乙酰化,提高p38的激酶活性,但HDAC3则使p38去乙酰化而抑制其活性[22]。心肌细胞特异性过表达的转基因小鼠的研究结果也显示,幼鼠心室心肌尤其是室间隔增厚、心室腔减小,但心肌增厚是由于心肌细胞增生而非心肌肥大;与转基因小鼠不同,转基因小鼠在3个月大时并未出现心肌肥大,也不增加ISO引起的心肌肥大程度[23]。
HDAC8也被证明可调节心肌肥大。在醋酸去氧皮质酮–盐诱导高血压的大鼠模型中,HDACs抑制剂丙戊酸钠处理使HDAC6和HDAC8的酶活性下降,抑制慢性高血压大鼠的心肌肥大及纤维化[24]。Yan等[25]研究显示miR-21-3p抑制TAC和血管紧张素(angiotensin IIAng II)诱导的心肌肥大的作用靶点是沉默HDAC8;HDAC8的表达通过提升磷酸化Akt和磷酸化Gsk3β而削弱miR-21-3p介导的心肌肥大抑制作用。Li等[20]研究也表明HDAC8和HDAC2在肾性高血压大鼠的心脏重塑中起关键作用。有关I类HDACs调控心肌肥大的相关作用机制见图2。
3 II类HDACs在心肌肥大中的作用
IIa类HDACs家族的调控能力主要通过在细胞核和细胞质之间穿梭来实现[53](图3)。IIa类HDACs家族具有较长的N-末端延伸,能够结合转录因子MEF2和伴侣蛋白14-3-3产生HDACs信号响应。IIa类HDACs家族成员被钙/钙调蛋白依赖性蛋白激酶(CaMK)或蛋白激酶D(PKD)等激酶磷酸化后与伴侣蛋白14-3-3结合,使IIa类HDACs与MEF2解聚,从细胞核穿梭至细胞质[54,55]。HAT p300则与游离的MEF2上的HDACs结合位点相结合,将MEF2从转录抑制状态转化为激活状态[56]。活化的MEF2转录因子通过直接促进肥大靶基因表达或与其他相关促肥大转录因子(如GATA和NFAT)相互作用促进肥大[57,58]。例如,在非经典Wnt/CaMKII信号途径中,Dvl1激活CaMKII信号磷酸化HDAC4,HDAC4通过保守的氨基末端α-螺旋结构域和HDAC5形成的复合物响应CaMKII信号促进HDAC5出核,激活MEF2转录因子的活性诱导心肌肥大[26]。而Dvl1引起的经典Wnt信号传导是通过形成β-catenin-TCF/ LEF复合物结合靶基因启动子,诱导Wnt信号的靶基因表达[59],且非经典Wnt/CaMKII信号与经典Wnt信号传导之间不相互影响。HDAC9敲除小鼠在出生8个月后出现心肌肥大现象,并且年幼的小鼠面对压力更容易出现心肌肥大现象。钙调神经磷酸酶信号磷酸化HDAC9,激活的HDAC9出核减少与MEF2形成转录阻遏物,对病理性肥大信号做出反应[34]。HDAC9的剪接变体MITR缺乏HDAC结构域,但与全长HDAC9蛋白一样可与MEF2在细胞核内相互作用形成转录阻遏物抑制MEF2的转录活性,调控心肌肥大[35]。
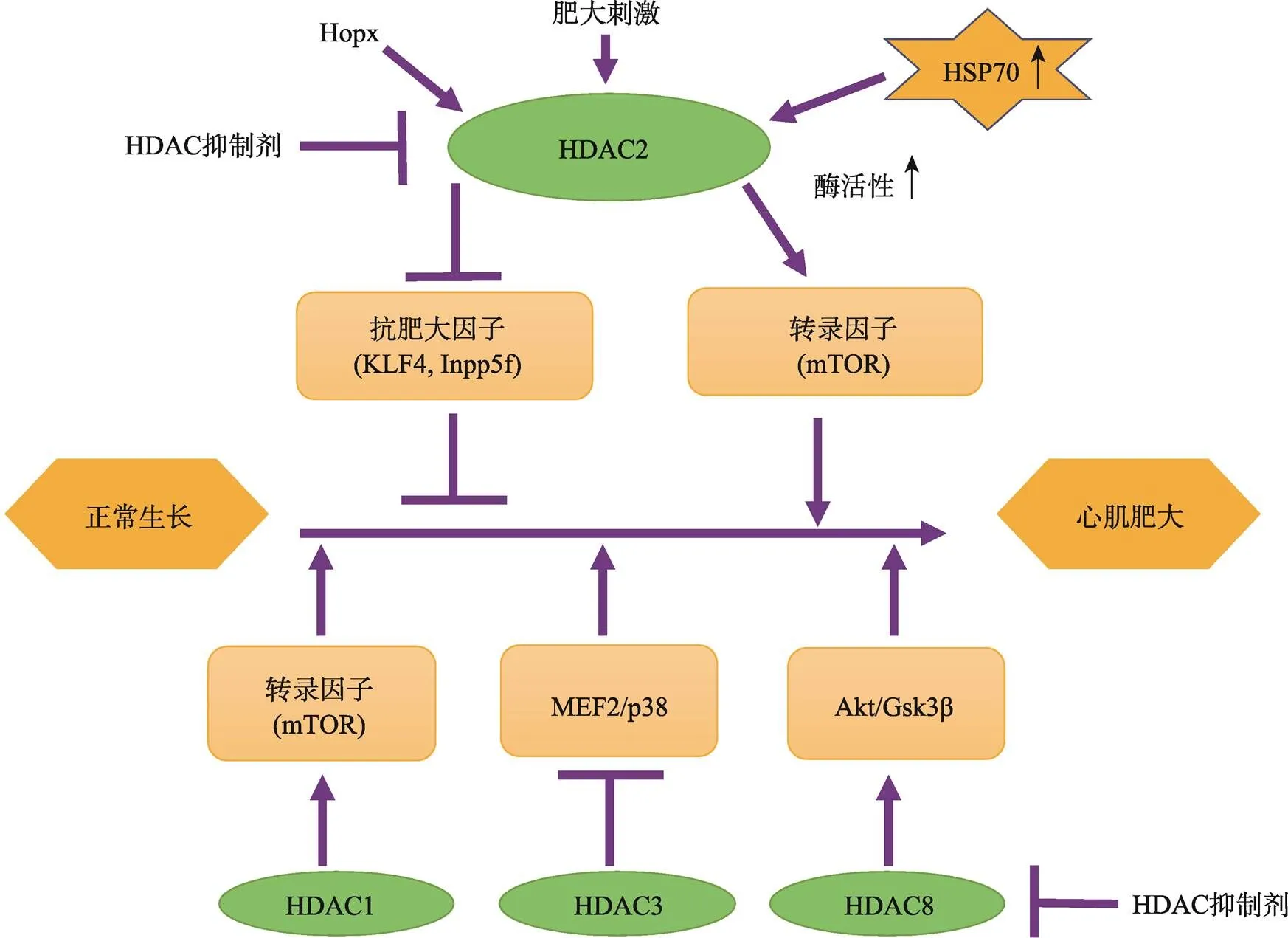
图2 I类HDACs调控心肌肥大的作用机制

图3 II类HDACs调控心肌肥大的作用机制
IIa类HDACs家族也可以结合促肥大转录因子,通过调控促肥大转录因子的活性进而抑制心肌细胞的肥大[60]。转录因子YY1可以抑制或激活多种基因的转录,其蛋白结构与磷酸化的HDAC5结构域结合。阻止HDAC5的核输出将抑制YY1的活性,进而下调肥大靶基因的表达,为抗病理性肥大提供保护机制[28]。IIa类HDACs能与Myocardin相互作用形成转录复合物,通过抑制Myocardin-SRF转录复合物与靶基因启动子CArG元件的结合下调SRF转录因子促肌生成的活性,抑制SRF调控的肥大靶基因表达[61,62]。CaMKII信号磷酸化HDAC4,通过去乙酰化作用降低MEF2和SRF转录因子的活性下调肥大靶标基因的表达[27]。
IIa类HDACs还可以通过发挥“酶”的功能抑制心肌肥大[63]。磷酸化的HDAC2是促进心肌肥大源动力,而HDAC2磷酸化的过程也受到IIa类HDACs乙酰化的影响,涉及到由PCAF /HDAC5介导的乙酰化和去乙酰化过程。HDAC5发挥去乙酰化酶活性去乙酰化肥大因子HDAC2,通过降低HDAC2乙酰化水平进而下调HDAC2的磷酸化水平抑制心肌肥大表型[29]。在心肌细胞中,HDAC5的催化活性能抑制线粒体活性氧(reactive oxygen species, ROS)的产生,通过下调NRF2转录因子调控的抗氧化剂基因的表达来应对外界强压引发的生理性心肌肥大[30]。
神经激素诱导心肌肥大关键机制就在IIa类HDACs从细胞核移位导致MEF2C的去阻遏和促肥大基因的活化。内皮素(endothelin, ET1)诱导的肥大机制一方面在于PKC/PKD的磷酸化HDAC5,通过诱导HDAC5出核降低对MEF2转录因子的阻遏促进心肌肥大;另一方面在于降低脑利钠肽、β-肌球蛋白重链、骨骼肌α-肌动蛋白、心肌肌动蛋等促肥大靶基因组蛋白H3去乙酰的表达水平[31]。ISO诱导的肥大机制一方面可以通过PKA信号和Gi-PI3K- Akt-CaMKII信号磷酸化IIa类HDACs,通过诱导IIa类HDAC出核激活MEF2信号下游肥大基因ANP和βMHC的表达[64,65];另一方面通过β-肾上腺素信号介导氧化过程,通过增加细胞质内的ROS激活CaMKII磷酸化HDAC5,通过诱导HDAC5出核激活MEF2转录因子的活性,此过程独立于PKD和PKA[32]。
IIb类HDACs中,HDAC10尚无报导其与心肌肥大有关,但HDAC6已被发现参与调节心肌肥大。如前文所述,HDAC6可能参与醋酸去氧皮质酮–盐诱导的慢性高血压大鼠的心肌肥大及纤维化[36]。尽管在注射Ang II后缺失HDAC6的小鼠与野生型小鼠一样发生了心脏肥大和纤维化,但是前者的心脏功能维持长达8周,而后者很快出现了收缩功能障碍。HDAC6的小分子抑制剂tubastatin A处理也呈现相似的表型。在TAC介导的压力超负荷情况下,HDAC6缺失型小鼠还表现出改善的左心室功能。HDAC6抑制似乎部分通过增强肌原纤维协调收缩而维持收缩功能[37]。大鼠动物实验的结果显示,口服丁酸钠可显著减轻Ang II诱导的大鼠心脏肥大反应,是通过以HDAC5/HDAC6依赖性方式抑制COX2/PGE2途径的激活实现的[33]。
4 III类HDACs在心肌肥大中的作用
III类HDACs是一类依赖于NADH的去乙酰化酶类家族,从细菌到人类高度保守,调节人类多种代谢、应激以及与衰老相关的过程,如糖尿病和心血管疾病等。心肌特异性敲除的小鼠出现糖尿病性心肌病症状,包括心脏肥大和功能障碍等缺陷、胰岛素抗性和葡萄糖代谢异常等[38]。SIRT6已被证明通过催化组蛋白去乙酰化来调控心肌肥大。在TAC手术或ISO注射后,基因敲除小鼠发展成心肌肥大和心力衰竭,而心肌特异性过表达的转基因小鼠不发生心肌肥大。SIRT6与c-Jun形成复合物而特异性募集到胰岛素样生长因子(insulin-like growth factor, IGF)信号相关基因启动子上,通过促使启动子中H3K9而非H3K14的去乙酰化,直接抑制IGF信号相关关键基因如Igf1r、Igf2r、Igf2、Akt1和Akt3等的转录,进而抑制IGF-Akt信号引起的心肌肥大和心力衰竭[49]。但是,目前的研究结果显示Ⅲ类HDACs调控心肌肥大的作用更多地通过调节非组蛋白的去乙酰化实现。SIRT1对介导与PIP3结合的Akt和PDK1的pleckstrin同源域的赖氨酸残基进行去乙酰化,增强了Akt和PDK1与PIP3的结合并促进了Akt的活化。这一翻译后修饰对于Akt的致癌作用以及其在体育锻炼和Ang II诱导的心肌肥大肥大中的作用都非常重要;敲除SIRT1基因对激动剂诱导的小鼠心肌肥大具有很强的保护作用[39]。还有有结果显示,在腹主动脉缩窄术或PE诱导的心肌肥大大鼠中,SIRT1可抑制PKC-ζ的乙酰化,阻止其与磷酸肌醇依赖性激酶1的结合,降低PKC-ζ磷酸化而下调NF-κB、ERK1/2和ERK5的活性来抑制心肌肥大[40]。FGF2既可以通过提高SIRT1的去乙酰基酶活性促进SIRT1 对LKB1的去乙酰化,进一步激活AMPK活性;也可通过提高SIRT1的去乙酰基酶活性改变FoxO1转录因子活性,上调过氧化氢酶、Sod2、Bim靶基因的表达,从而减少ROS积累和心肌细胞的凋亡,缓解Ang II引起的心肌肥大[41]。化疗药物阿霉素(Doxo)会导致ROS产生增多、线粒体片段化,诱发心脏线粒体损伤和细胞死亡而引发心肌肥大。SIRT3的激活可保护线粒体免受损伤,从而保护心脏免受Doxo诱导的心脏毒性的影响[43]。基因敲除和转基因的小鼠模型的研究结果显示,SIRT3是通过抑制细胞内ROS水平来保护心脏免于发生心脏肥大和间质纤维化的。SIRT3和Foxo3a能够在体内相互结合,以NAD依赖的方式使Foxo3a去乙酰化,掩盖Foxo3a的磷酸化依赖性核输出,使其在核内不动,从而促进依赖于Fox3a的锰超氧化物歧化酶(MnSOD)和过氧化氢酶(Cat)的编码基因转录,降低细胞内ROS的水平,再通过MAPK/ERK和PI3K/Akt途径抑制了Ras激活和其下游信号传导,导致参与心脏肥大发展的转录因子(特别是GATA4和NFAT)、翻译因子(特别是elf4E)以及核糖体蛋白S6P的活性受到抑制,从而抑制心肌肥大[44]。在基因敲除和转基因小鼠模型中,外源NAD的抗肥大作用是通过激活SIRT3而不是SIRT1来介导的。SIRT3使LKB1脱乙酰基并激活,从而增强LKB1-AMPK途径的活性,阻止了mTOR介导的蛋白质合成[45]。SIRT2通过使激酶LKB1的48赖氨酸脱乙酰化从而促进LKB1的磷酸化并随后激活AMPK而导致LKB1-AMPK信号传导,敲除则降低AMPK的激活,促进衰老相关和Ang II引起的心脏肥大[42]。SIRT4表现为提高ROS水平促进心肌肥大。在Ang II刺激诱导的转基因肥大小鼠模型中,SIRT4抑制线粒体中MnSOD与SIRT3结合,通过增加MnSOD乙酰化水平降低其活性,导致ROS积累进而促进肥大[46]。SIRT5通过对蛋白的去琥珀酰化作用调控代谢抑制心肌肥大。SIRT5可调控赖氨酸去琥珀酰化,在基因敲除小鼠心脏中SIRT5的缺失导致ECHA酶的赖氨酸琥珀酰化而降低活性,出现脂肪酸代谢缺陷和ATP含量降低现象,并产生肥厚型心肌病[47]。在TAC引起的心肌肥大的模型中,SIRT5参与细胞氧化代谢的蛋白质底物(丙酮酸脱氢酶和脂肪酸氧化酶)的去琥珀酰化影响抑制葡萄糖氧化、TCA循环、脂肪酸氧化,通过调控线粒体能量来应对心脏的应激反应[48]。在心肌细胞特异性敲除基因的小鼠模型中,小鼠心脏肥大、心脏的收缩功能明显降低。SIRT7通过与GATA4直接相互作用并促进其脱乙酰基、降低GATA4的转录活性而发挥抗肥大作用[50]。
多种miRNA靶向HDACs在心肌肥大中起重要的调控作用。miR-21-3p抑制心肌肥大的作用靶点是HDAC8,在TAC和Ang II引起的心肌肥大模型中,下调的HDAC8降低Akt和Gsk3β磷酸化抑制肥大[25]。miR-22是心肌肥大和心脏重塑的关键调节因子,在TAC诱导心肌肥大的小鼠中,敲除miR-22促进靶基因SERCA2a等的表达,或通过促进PURB抑制SRF转录因子的活性,进而抑制心肌肥大和心力衰竭[66]。反之,过表达miR-22则诱导心肌肥大,其中和被鉴定为miR-22下游靶标基因[67]。miR-1可能通过靶向HADC4在甲状腺激素诱导的心肌肥大中起作用。在新生大鼠心肌细胞以及Wistar大鼠中,甲状腺激素刺激诱导心肌肥大时,miR-1的下调,同时伴随HDAC4 表达增高;过表达miR-1可阻止新生大鼠心肌细胞中T3诱导的心肌肥大和HADC4 mRNA水平的下降;另外HDACs抑制剂处理则能有效抑制心肌肥大[68]。在Ang II和TAC引起的心肌肥大的细胞模型和小鼠模型中,miR-497的表达明显降低,萤光素酶报告基因检测结果表明是miR-497的直接靶基因[69]。
5 HDACs抑制剂的抗心肌肥大作用
目前,针对HDACs家族为靶标的HDACs抑制剂在心肌肥大的治疗上获得有效研究进展。广谱HDAC抑制剂TSA在Ang II或主动脉束带引起的心肌肥大小鼠/大鼠模型中, 可降低心脏中ANP、α-tubulin、β-MHC和间质纤维化基因的表达,明显减弱心肌肥大现象,提高鼠的存活率[70]。TSA也能减轻异丙肾上腺素[71]、苯肾上腺素[71,72]诱导的心肌细胞肥大反应。在IL-18诱导的小鼠心肌肥大模型中,TSA通过促进PTEN-PI3K-AKT和MAPK信号通路中激酶(TGFβ、TNF-α、IFNγ和IL-6)及转录因子(Myc、p53、NFkB和HNF4A)的乙酰化来改善心肌肥大现象[73,74]。在Ang II诱导的心肌肥大小鼠/大鼠模型中,TSA可通过抑制HDAC6活性而减轻心肌肥大反应[33,37]。在Hop转基因小鼠心肌肥大模型中,TSA通过抑制依赖HDAC2的SRF活性减轻心肌肥大反应[75]。广谱HDAC抑制剂丁酸钠能够有效减少苯肾上腺素诱导诱导的心肌细胞的肥大反应[72]以及Ang II诱导的小鼠/大鼠心肌肥大反应[33,37]。scriptaid[76]、CBHA[73,74]也显示具有TSA类似的抗心肌肥大作用。HDACs抑制剂SAHA可以减轻醋酸去氧皮质酮-盐诱导高血压大鼠模型中相关的心血管的重塑,并改善心脏和血管的心血管结构和功能(包括心室肥大)[77]。一些选择性HDAC抑制剂也有抑制心肌肥大的效应。I类HDAC抑制剂丙戊酸或SK-7041可减轻Ang II或主动脉束带引起的小鼠/大鼠心肌肥大[70]。还有研究显示丙戊酸通过减少心脏中的ROS和ANG II1型受体的表达及对盐皮质激素受体MR乙酰化作用来减轻肥大反应,减弱自发性高血压大鼠心脏的肥大和纤维化反应[78]。丙戊酸也能通过抑制Hop转基因小鼠的心肌肥大反应[75]。丙戊酸钠能有效阻断肺动脉束带或单芥子碱注射诱导的大鼠右心室肥大[79],以及醋酸去氧皮质酮–盐诱导的慢性高血压大鼠的心肌肥大及纤维化反应[24,80]。另一种I类HDAC抑制剂Apicidin可有效减轻主动脉束诱导的小鼠左室肥大和心脏纤维化现象,改善心脏收缩功能[81]。在不同的心肌肥大动物模型中施用HDACs抑制剂治疗心肌肥大取得了良好的效果,特别是泛抑剂TSA已经有了一些临床应用报告[76,82]。但是HDAC抑制剂在人类心肌肥大治疗中会伴随出现白细胞减少、贫血、骨髓抑制等副作用,且临床研究数据较少,所以距离HDAC抑制剂在治疗人类心肌肥大方面的应用还需要更多的临床应用研究。
6 结语与展望
HDACs家族在心脏的发育和心肌肥大的过程中扮演着重要的角色。其中I类HDACs和IIa类HDACs、III类HDACs在调控心肌肥大中发挥着不同的作用。I类HDACs通过直接下调抗肥大调节因子而促进心肌肥大,或通过调控转录因子和酶活性调控心肌肥大;IIa类HDACs通过细胞核和细胞质之间穿梭减弱对MEF2转录因子活性的抑制,或通过抑制促肥大转录因子活性,或发挥去乙酰化酶作用抑制心肌细胞的肥大;III类HDACs则通过调控肥大信号途径、氧化代谢途径的酶和转录因子活性,或发挥去琥珀化酶功能等多种途径调控心肌肥大。虽然I类HDACs在心肌肥大过程中起着重要的作用,但IIa类HDACs以多形式发挥着抑制心肌肥大的作用, 而近几年III类HDACs则通过氧化代谢途径作用调控心肌肥大也成为研究热点。HDACs抑制剂除了在治疗肿瘤等代谢性疾病方面具有潜在的应用价值[83],目前研究表明一些HDAC抑制剂尤其是一些广谱HDAC抑制剂已经在阻止心肌肥大、纤维化、局部心肌缺血显示出明显的效果。
综上所述,大量的研究证明HDACs在心肌肥大病理过程中发挥了重要的调节作用。虽然HDACs在体内的作用靶点、参与不同肥大刺激因子诱导心肌肥大过程中的具体HDACs的作用和相互关系等问题还有待进一步解析,调控通路尚未完全阐明,与HDACs直接相互作用的其他心肌肥大调控因子还需要继续鉴定,各种HDAC抑制剂的作用靶标和精确效应有待精细化研究,HDAC抑制剂的抑制效应与靶HDAC的基因敲除表型之间的差异及其原因等重要问题急待更深入的研究解决,但HDACs极有可能成为对心肌肥大和心脏重塑进行有效临床干预的新治疗策略。
[1] Hunter DJ, Reddy KS. Noncommunicable diseases., 2013, 369(14): 1336–1343.
[2] Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation?, 2000, 19(6): 1176–1179.
[3] Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p., 1996, 272(5260): 408–411.
[4] Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, Rowley M, Serafini S, Steinkühler C. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases., 2008, 18(6): 1814–1819.
[5] Duan BY, Ye D, Zhu SC, Jia WW, Lu CQ, Wang GY, Guo XD, Yu YY, Wu CY, Kang JH. HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ ERK axis., 2017, 8(37): 61338–61349.
[6] Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan., 2012, 13(4): 225–238.
[7] Cao J, Sun L, Aramsangtienchai P, Spiegelman NA, Zhang XY, Seto E, Lin HN. DAC11 regulates type I interferon signaling through defatty-acylation of SHMT2., 2019, 116(12): 5487–5492.
[8] Morales CR, Li DL, Pedrozo Z, May HI, Jiang N, Kyrychenko V, Cho GW, Kim SY, Wang ZV, Rotter D, Rothermel BA, Schneider JW, Lavandero S, Gillette TG, Hill JA. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression., 2016, 9(422): ra34.
[9] Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression., 2002, 21(11): 2672–2681.
[10] Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression., 2003, 13(2): 143–153.
[11] Kesherwani V, Nandi SS, Sharawat SK, Shahshahan HR, Mishra PK. Hydrogen sulfide mitigates homocysteine- mediated pathological remodeling by inducing miR-133a in cardiomyocytes., 2015, 404(1–2): 241–250.
[12] Chandrasekaran S, Peterson RE, Mani SK, Addy B, Buchholz AL, Xu L, Thiyagarajan T, Kasiganesan H, Kern CB, Menick DR. Histone deacetylases facilitate sodium/ calcium exchanger up-regulation in adult cardiomyocytes., 2009, 23(11): 3851–3864.
[13] Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi XX, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility., 2007, 21(14): 1790–1802.
[14] Yuan YG, Peng WZ, Liu YX, Xu ZS. Palmatine attenuates isoproterenol-induced pathological hypertrophyselectively inhibiting HDAC2 in rats., 2017, 30(4): 406–412.
[15] Trivedi CM, Luo Y, Yin Z, Zhang MZ, Zhu WT, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity., 2007, 13(3): 324–331.
[16] Eom GH, Kook H. Role of histone deacetylase 2 and its posttranslational modifications in cardiac hypertrophy., 2015, 48(3): 131–138.
[17] Raghunathan S, Goyal RK, Patel BM. Selective inhibition of HDAC2 by magnesium valproate attenuates cardiac hypertrophy., 2017, 95(3): 260– 267.
[18] Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, Zhou R, Ferrari V, Gruber P, Epstein JA. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop., 2003, 112(6): 863–871.
[19] Trivedi CM, Zhu W, Wang Q, Jia C, Kee HJ, Li L, Hannenhalli S, Epstein JA. Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation., 2010, 19(3): 450–459.
[20] Li RF, Cao SS, Fang WJ, Song Y, Luo XT, Wang HY, Wang JG. Roles of HDAC2 and HDAC8 in cardiac remodeling in renovascular hypertensive rats and the effects of valproic acid sodium., 2017, 99(1–2): 27–39.
[21] Grégoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2., 2007, 27(4): 1280–95.
[22] Pillai VB, Sundaresan NR, Samant SA, Wolfgeher D, Trivedi CM, Gupta MP. Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes., 2011, 31(11): 2349–2363.
[23] Trivedi CM, Lu MM, Wang QH, Epstein JA. Transgenic over-expression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy., 2008, 283(39): 26484–26489.
[24] Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, Jeong MH. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive ratsregulation of HDAC6/HDAC8 enzyme activity., 2013, 37(4–5): 229–239.
[25] Yan MW, Chen C, Gong W, Yin ZW, Zhou L, Chaugai S, Wang DW. miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8., 2015, 105(3): 340–352.
[26] Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN. Histone deacetylase 5 acquires calcium/calmodulin- dependent kinase II responsiveness by oligomerization with histone deacetylase 4., 2008, 28(10): 3437–3445.
[27] Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, Olson EN. Selective repression of MEF2 activity by PKA- dependent proteolysis of HDAC4., 2011, 195(3): 403–415.
[28] Sucharov CC, Dockstader K, McKinsey TA. YY1 protects cardiac myocytes from pathologic hypertrophy by interacting with HDAC5., 2008, 19(10): 4141–4153.
[29] Eom GH, Nam YS, Oh JG, Choe N, Min HK, Yoo EK, Kang G, Nguyen VH, Min JJ, Kim JK, Lee IK, Bassel- Duby R, Olson EN, Park WJ, Kook H. Regulation of acetylation of histone deacetylase 2 by p300/CBP-Associated Factor/Histone deacetylase 5 in the development of cardiac hypertrophy., 2014, 114(7): 1133–1143.
[30] Hu T, Schreiter FC, Bagchi RA, Tatman PD, Hannink M, McKinsey TA. HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression., 2019, 294(21): 8640–8652.
[31] Zhang H, Shao Z, Alibin CP, Acosta C, Anderson HD. Liganded peroxisome proliferator-activated receptors (ppars) preserve nuclear histone deacetylase 5 levels in endothelin-treated sprague-dawley rat cardiac myocytes, 2014, 9(12): e115258.
[32] Haworth RS, Stathopoulou K, Candasamy AJ, Avkiran M. Neurohormonal regulation of cardiac histone deacetylase 5 nuclear localization by phosphorylation-dependent and phosphorylation-independent mechanisms., 2012, 110(12): 1585–1595.
[33] Zhang LL, Deng MK, Lu AH, Chen YT, Chen Y, Wu C, Tan Z, Boini KM, Yang TX, Zhu Q, Wang L. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathwaya HDAC5/HDAC6-dependent mechanism., 2019, 23(12): 8139–8150.
[34] Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal- responsive repressors of cardiac hypertrophy., 2002, 110(4): 479–488.
[35] Zhang CL, McKinsey TA, Olson EN. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis., 2001, 98(13): 7354– 7359.
[36] Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, Jeong MH. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive ratsregulation of HDAC6/HDAC8 enzyme activity., 2013, 37(4–5): 229–239.
[37] Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, Schuetze KB, Horn TR, Chen B, Ferrara C, Scellini B, Piroddi N, Tesi C, Poggesi C, Jeong MY, McKinsey TA. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling., 2014, 307(2): H252–258.
[38] Ma S, Feng J, Zhang R, Chen JW, Han D, Li X, Yang B, Li XJ, Fan MM, Li CY, Tian ZH, Wang YB, Cao F. SIRT1 activation by resveratrol alleviates cardiac dysfunctionmitochondrial regulation in diabetic cardiomyopathy mice., 2017, 2017: 4602715.
[39] Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase sirt1 promotes membrane localization and activation of akt and pdk1 during tumorigenesis and cardiac hypertrophy., 2011, 4(182): ra46.
[40] Li JY, Huang JY, Lu J, Guo Z, Li ZM, Gao H, Wang PX, Luo WW, Cai SD, Hu YH, Guo KT, Wang LP, Li ZZ, Wang MH, Zhang XL, Liu PQ. Sirtuin 1 represses PKC-ζ activity through regulating interplay of acetylation and phosphorylation in cardiac hypertrophy., 2019, 176(3): 416–435.
[41] Li ST, Zhu ZX, Xue M, Yi XC, Liang JJ, Niu C, Chen G, Shen YJ, Zhang HP, Zheng JY, Zhao CC, Liang YZ, Cong WT, Wang Y, Jin LT. Fibroblast growth factor 21 protects the heart from angiotensin II-induced cardiac hypertrophy and dysfunctionSIRT1., 2019, 1865(6): 1241–1252.
[42] Tang XQ, Chen XF, Wang NY, Wang XM, Liang ST, Zheng W, Lu YB, Zhao X, Hao DL, Zhang ZQ, Zou MH, Liu DP, Chen HZ. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy., 2017, 136(21): 2051–2067.
[43] Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S, Gupta MP. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin- induced cardiomyopathy in mice., 2016, 310(8): H962–972.
[44] Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting foxo3a-dependent antioxidant defense mechanisms in mice, 2009, 119(9): 2758–2771.
[45] Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous nad blocks cardiac hypertrophic response via activation of the sirt3-lkb1-amp-activated kinase pathway., 2010, 285(5): 3133–3144.
[46] Luo YX, Tang XQ, An XZ, Xie XM, Chen XF, Zhao X, Hao DL, Chen HZ, Liu DP. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity., 2017, 38(18): 1389–1398.
[47] Sadhukhan S, Liu XJ, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, Auwerx J, Lin HN. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function., 2016, 113(16): 4320– 4325.
[48] Hershberger KA, Abraham DM, Martin AS, Mao L, Liu J, Gu H, Locasale JW, Hirschey MD. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload., 2017, 292(48): 19767–19781.
[49] Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, Gupta MP. The sirtuin sirt6 blocks igf-akt signaling and development of cardiac hypertrophy by targeting c-jun., 2012, 18(11): 1643–1650.
[50] Yamamura S, Izumiya Y, Araki S, Nakamura T, Kimura Y, Hanatani S, Yamada T, Ishida T, Yamamoto M, Onoue Y, Arima Y, Yamamoto E, Sunagawa Y, Yoshizawa T, Nakagata N, Bober E, Braun T, Sakamoto K, Kaikita K, Morimoto T, Yamagata K, Tsujita K. Cardiomyocyte Sirt (Sirtuin) 7 ameliorates stress-induced cardiac hypertrophy by interacting with and deacetylating GATA4., 2020, 75(1): 98–108.
[51] Schueler M, Zhang Q, Schlesinger J, Tönjes M, Sperling SR. Dynamics of Srf, p300 and histone modifications during cardiac maturation in mouse., 2012, 8(2): 495–503.
[52] Schlesinger J, Schueler M, Grunert M, Fischer JJ, Zhang Q, Krueger T, Lange M, Tönjes M, Dunkel I, Sperling SR. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs., 2011, 7(2): e1001313.
[53] Mathias RA, Guise AJ, Cristea IM. Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease., 2015, 14(3): 456–470.
[54] Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5., 2004, 24(19): 8374–8385.
[55] McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation., 2000, 408(6808): 106–111.
[56] Sparrow DB, Miska EA, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun TJ. MEF-2 function is modified by a novel co-repressor, MITR., 1999, 18(18): 5085–5098.
[57] Morin S1, Charron F, Robitaille L, Nemer M. GATA- dependent recruitment of MEF2 proteins to target promoters., 2000, 19(9): 2046–2055.
[58] Youn HD, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis., 2000, 19(16): 4323–4331.
[59] Zhang M, Hagenmueller M, Riffel JH, Kreusser MM, Bernhold E, Fan J, Katus HA, Backs J, Hardt SE. Calcium/Calmodulin-dependent protein kinase II couples Wnt signaling with histone deacetylase 4 and mediates dishevelled-induced cardiomyopathy., 2015, 65(2): 335–344.
[60] Calalb MB, McKinsey TA, Newkirk S, Huynh K, Sucharov CC, Bristow MR. Increased phosphorylation- dependent nuclear export of class II histone deacetylases in failing human heart., 2009, 2(5): 325–332.
[61] Cao DS, Wang ZG, Zhang CL, Oh J, Xing W, Li S, Richardson JA, Wang DZ, Olson EN. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin., 2005, 25(1): 364–376.
[62] McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression., 2006, 116(1): 36–48.
[63] Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR., 2002, 9(1): 45–57.
[64] Chang CW, Lee L, Yu D, Dao K, Bossuyt J, Bers DM. Acute β-adrenergic activation triggers nuclear import of histone deacetylase 5 and delays G(q)-induced transcriptional activation., 2013, 288(1): 192–204.
[65] Mangmool S, Shukla AK, Rockman HA. β-arrestin- dependent activation of Ca2+/Calmodulin kinase Ⅱ after β-adrenergic receptor stimulation., 2010, 189(3): 573–587.
[66] Gurha P, Abreu-Goodger C, Wang T, Ramirez MO, Drumond AL, van Dongen S, Chen Y, Bartonicek N, Enright AJ, Lee B, Kelm RJ Jr, Reddy AK, Taffet GE, Bradley A, Wehrens XH, Entman ML, Rodriguez A. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction., 2012, 125(22): 2751–2761.
[67] Huang ZP, Chen JH, Seok HY, Zhang Z, Kataoka M, Hu XY, Wang DZ. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress., 2013, 112(9): 1234–1243.
[68] Diniz GP, Lino CA, Moreno CR, Senger N, Barreto-Chaves MLM. MicroRNA-1 overexpression blunts cardiomyocyte hypertrophy elicited by thyroid hormone., 2017, 232(12): 3360–3368.
[69] Xiao Y, Zhang X, Fan S, Cui G, Shen Z. MicroRNA-497 inhibits cardiac hypertrophy by targeting Sirt4., 2016, 11(12): e0168078.
[70] Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding., 2006, 113(1): 51–59.
[71] Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy., 2011, 108(10): 4123–4128.
[72] Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors., 2003, 278(31): 28930–28937.
[73] Majumdar G, Adris P, Bhargava N, Chen H, Raghow R. Pan-histone deacetylase inhibitors regulate signaling pathways involved in proliferative and pro-inflammatory mechanisms in H9c2 cells., 2012, 13: 709.
[74] Majumdar G, Rooney RJ, Johnson IM, Raghow R. Panhistone deacetylase inhibitors inhibit proinflammatory signaling pathways to ameliorate interleukin-18-induced cardiac hypertrophy., 2011, 43(24): 1319–1333.
[75] Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, Zhou R, Ferrari V, Gruber P, Epstein JA. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop., 2003, 112(6): 863–871.
[76] Kong YQ, Tannous P, Lu GR, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy., 2006, 113(22): 2579–2588.
[77] Iyer A, Fenning A, Lim J, Le GT, Reid RC, Halili MA, Fairlie DP, Brown L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats., 2010, 159(7): 1408–1417.
[78] Kang SH, Seok YM, Song MJ, Lee HA, Kurz T, Kim I. Histone deacetylase inhibition attenuates cardiac hypertrophy and fibrosis through acetylation of mineralocorticoid receptor in spontaneously hypertensive rats., 2015, 87(5): 782–791.
[79] Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI, Ma JS, Kook H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats., 2010, 74(4): 760–770.
[80] Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM, Francis J. HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats., 2010, 56(3): 437–444.
[81] Gallo P, Latronico MV, Gallo P, Grimaldi S, Borgia F, Todaro M, Jones P, Gallinari P, De Francesco R, Ciliberto G, Steinkühler C, Esposito G, Condorelli G. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure., 2008, 80(3): 416–424.
[82] Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, El-Osta A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes., 2015, 10(5): 418–430.
[83] Cao JH, Liao WT, Wo C,Xu GR, Xu HX, Li PL, Tao Y, Wang P, Lin JR, Deng LR. Transcriptome screening and verification of genes related to metabolism affected by histone deacetylase inhibitors., 2015, 37(9): 918–925.曹继红, 廖尉廷, 沃琤, 徐国荣, 徐焕新, 李平龙, 陶冶, 王鹏, 林加日, 邓连瑞. 组蛋白去乙酰化酶抑制剂影响的代谢相关基因的组学筛查及验证. 遗传, 2015, 37(9): 918–925.
The mechanism underlying histone deacetylases regulating cardiac hypertrophy
Lian Ren, Xiushan Wu, Yongqing Li
Cardiac hypertrophy is a compensatory response that occurs as a result of increased hemodynamic requirement in peripheral tissues. In the process of cardiac hypertrophy, the expression of different types of genes in different stages is transcriptionally regulated by multiple-level physiological and pathological signals. Histone acetylation, as the most extensive post-translational modification, is closely controlled by the antagonistic histone acetyltransferases (HAT) and histone deacetylases (HDACs). Recent studies have shown that HDACs, as a family of enzymes that inhibit transcription and contain highly conserved deacetylase domains, regulate gene expression during cardiac hypertrophy through a variety of pathways. In this review, we mainly summarize the research progress on histone deacetylase in cardiac hypertrophy. By elucidating the role and molecular mechanism of different HDACs in cardiac hypertrophy, it provides new ideas for the treatment of different types of cardiac hypertrophy and heart failure, and molecular targets for new drug design.
myocardial hypertrophy; post-translational modification; transcription process; histone deacetylases; molecular mechanism
2020-01-18;
2020-04-13
国家自然科学基金项目(编号:81470377)和湖南省生物发育工程及新产品研发协同创新中心项目(编号:2013-448-6)资助[Supported by the National Natural Science Foundation of China (No. 81470377), and Hunan Province Biological Development Engineering and New Product R & D Collaborative Innovation Center (No. 2013-448-6)]
任恋,在读博士研究生,专业方向:分子遗传。E-mail: 82287168@qq.com
李永青,博士,教授,研究方向:心脏发育与疾病的分子机制。E-mail: liyongqing2002cn@aliyun.com
10.16288/j.yczz.19-346
2020/4/28 8:44:03
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20200427.1716.003.html
(责任编委: 杨中州)
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
中成药(2017年8期)2017-11-22
作文教学研究(2016年1期)2016-07-05
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年8期)2015-06-22
