
获得性低促性腺激素性性腺功能减退症临床分析(附6例报告)
2020-06-29 09:11
青岛大学学报(医学版) 2020年3期
(中国人民解放军海军第971医院内分泌科,山东 青岛 266071)
低促性腺激素性性腺功能减退症(HH)是由于先天性或获得性等多种病因导致下丘脑异常,生成促性腺激素(GnRH)和(或)垂体生成黄体生成素(LH)和(或)促卵泡刺激素(FSH)不足,阻碍正常的睾丸/卵巢分泌功能的一类疾病[1]。男女均可发病,男性发病率高于女性,男女之比约为5∶1[2]。HH按病因可分为特发性HH(IHH)和获得性HH(AHH)。随着鞍区CT及MRI检查的发展普及,AHH病人多能找到明确的病因,主要包括下丘脑区及垂体区肿瘤、手术、放疗损伤等,也可以是某些全身疾病在内分泌系统的表现。目前,对IHH的临床研究报道较多,但对AHH的研究甚少。为提高对AHH的认识,早期诊断和治疗该病,现将我科收治的6例AHH病人的临床资料报告如下。
1 临床资料
1.1 一般资料
2010年1月—2019年7月,我科收治AHH住院病人共6例,男5例,女1例;发病年龄为23~49岁,中位发病年龄为36.3(25.4,47.2)岁;病程为1~30年,平均为10.2年;体质量指数(BMI)为(22.3±11.5)kg/m2。AHH的病因:遗传性血色病1例,颅咽管瘤术后3例,生长激素瘤术后1例,无功能垂体大腺瘤术后1例。
1.2 临床表现
本组6例AHH病人,1例以性功能减退为首发症状,余5例均以视物模糊、视力下降为首发症状。其中1例起病时视力下降同时合并肢端肥大;1例反复腹泻2年后出现视力下降至失明,垂体瘤切除术后2次复发,行伽马刀放疗后出现嗅觉丧失,并多次出现癫痫发作。1例男性病人第二性征发育不全,伴颜面部皮肤及手背色素沉着;余4例男性病人睾丸及第二性征发育正常。1例女性病人乳房及第二性征发育正常。6例病人均存在性功能减低和骨质疏松症,5例垂体和鞍区肿瘤术后病人均合并继发性甲状腺功能减退症和肾上腺功能减退症,3例合并中枢性尿崩症。
本组6例病人的性激素、GnRH水平均低于正常。其中1例病人生化指标明显高于正常:谷丙转氨酶(ALT)79 U/L,谷草转氨酶(AST)59 U/L,血清铁48.2 μmol/L,铁蛋白大于2 000 μg/L;GnRH兴奋试验和GnRH兴奋延迟试验结果提示垂体GnRH储备不足;肝脏CT检查示肝密度明显增高(白肝),提示早期肝硬化;肝脏MRI检查显示肝实质与胰腺T1WI及T2WI呈弥漫性明显低信号(黑肝);肝脏穿刺活组织病理检查(苏木精-伊经染色)示肝细胞内弥漫性含铁血黄素沉积,形态符合含铁血黄素沉着症(血色病);基因检测显示HFE2基因(HJV基因)突变,1~4号外显子杂合缺失,并且已经过MLPA方法证实(表1)。诊断为遗传性血色病(ⅡA型)。
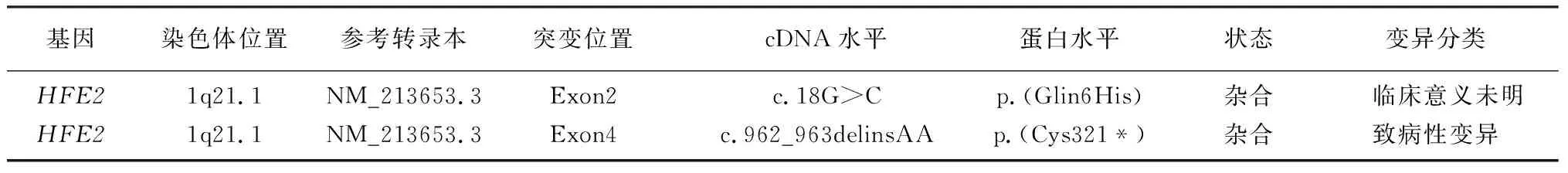
表1 遗传性血色病病人基因检测结果
1.3 治疗方法
遗传性血色病病人自行限制含铁食物的摄取,因地拉罗司及去铁胺等铁螯合剂青岛地区未采购到,且病人拒绝行放血治疗,给予口服十一酸睾酮替代及保肝药物治疗,3个月后病人第二性征和性功能有所好转,复查性腺激素水平较前升高但仍低于正常。其余5例病人,3例行经单鼻蝶窦占位切除术,2例行经翼点入路颅咽管瘤切除术,其中1例垂体瘤复发行开颅手术。术后病理检查示垂体瘤2例(无功能垂体大腺瘤和生长激素瘤各1例),颅咽管瘤3例。该5例病人均于术后1~3个月行辅助放疗,放疗剂量(12~40) Gy/(4~6)周。病人术后或放疗后均接受内分泌治疗。5例垂体和鞍区肿瘤病人因同时合并继发性甲状腺功能减退症和继发性肾上腺功能减退症,给予泼尼松和左甲状腺素钠替代治疗。1例老年女性病人给予泼尼松治疗后多次出现低钠血症,换用氢化可的松后低钠血症未再出现。5例男性病人均给予性激素替代治疗(口服十一酸睾酮胶丸40~80 mg,每天1~2次),性功能及肌力部分改善;老年女性病人考虑年龄因素,未行性激素替代治疗。6例病人因存在继发性骨质疏松,均给予补钙和活性维生素D治疗。
2 讨 论
下丘脑、垂体、性腺共同构成的生殖调节系统是青春期启动以及性腺分泌性激素的核心环节,所有威胁到下丘脑-垂体-性腺轴(HPG)的病变均会造成性腺相关激素的合成和分泌障碍,导致性腺功能减退症的发生。性腺功能减退症病人的临床特征主要表现为缺乏第二性征、性欲减退、生育功能障碍等。本文6例病人中,遗传性血色病病人发病年龄较小,表现为类无睾体型,第二性征虽有发育,但发育不完全,生殖器发育也不完全,考虑垂体功能减退症是随铁沉积累积效应逐渐发生的;其余病人发病年龄在28~49岁,年龄偏大,且合并多种垂体激素缺乏,考虑主要致病原因是垂体和鞍区肿瘤。
血色病又称含铁血黄素沉着症,是一种罕见的遗传性铁代谢性疾病,病人肠道铁吸收的不适当增加导致过量的铁在实质性细胞中沉积,最终导致组织损伤和多种器官功能受损。该病根据病因可分为原发性和继发性两大类。原发性血色病(PHC)又称特发性或遗传性血色病[3],为常染色体遗传性铁代谢疾病[4-5]。其常见的临床表现为心肌病、肝硬化、关节炎、皮肤色素沉着、内分泌失调如糖尿病、HH、甲状腺功能减退症等[6-9]。PHC根据基因突变位点及表型的不同,可分为Ⅰ型(HFE)[10]、ⅡA型(HJV)、ⅡB型(HAMP)、Ⅲ型(TFR2)和Ⅳ型(SLC40A1)[11]。Ⅰ~Ⅲ型均为常染色体隐性遗传,Ⅳ型为常染色体显性遗传[12-13]。Ⅱ型遗传性血色病又称青少年型遗传性血色病,是PHC中最严重的类型,一般在30岁前发病[14]。ⅡA型是最常见的PHC类型,其最常见的临床表现是性腺功能低下,其次是铁超负荷心肌病(最常见的死亡原因)和肝硬化[15-16]。性腺功能减退症在两性中均可发生,并可能早于其他临床症状出现。其临床表现包括性欲减退、阳痿、闭经、睾丸萎缩、男性乳房发育和体毛稀少等,上述改变的原因主要是铁沉积损害下丘脑-垂体功能所致的GnRH产生减少;肾上腺功能不全、甲状腺功能减退症和甲状旁腺功能减退症少见[17]。HH是血色病的一种重要的并发症[18],而血色病其他垂体轴功能通常正常,表明铁对促性腺细胞有亲和力[19-22]。PEDERSEN-BJERGAARD等[23]对血色病病人垂体功能的综述表明,GnRH缺乏伴临床性腺功能减退症的患病率为46%;除此之外,他们还报道了其他轴的功能减退,如生长激素缺乏占受试者的15%,泌乳素缺乏占8%,促甲状腺素缺乏占4%,促肾上腺皮质激素缺乏占1.5%,泌乳素、促甲状腺激素、促肾上腺皮质激素的缺乏通常与性腺功能减退或生长激素缺乏同时存在。本文遗传性血色病病人主要表现为单纯性HH,其他垂体轴无累及。另外,该病人还表现为肝硬化、糖代谢异常、皮肤色素沉着等。肝脏的CT和MRI检查分别呈现“白肝”和“黑肝”的特异性表现,同时垂体MRI检查表现为T2WI信号明显减低,此为铁沉积在MRI的T2WI上的特异性表现[24],为临床医生识别血色病提供了途径。血色病导致的HH除治疗原发病外,同时应给予性激素替代治疗,改善糖脂代谢、肌肉力量及性功能。有生育要求时可联合HCG+HMG生精治疗,以提高病人的生活质量。
本文其余5例均为垂体和鞍区肿瘤术后病人。垂体和鞍区肿瘤为神经科临床常见的肿瘤,多数需行手术治疗。目前除极少数病人需要开颅手术切除肿瘤外,绝大多数病人的垂体和鞍区肿瘤可采用经鼻蝶入路手术切除[25-28]。上述方法在切除肿瘤、改善生化指标、缓解原发症状及减少并发症等方面均表现出积极的效果[29-30]。手术治疗后的急性严重并发症如颅内感染、脑脊液鼻漏、视神经损伤、癫痫等多能及时发现并处理,但下丘脑-垂体损伤导致的内分泌激素异常以及由此带来的一系列潜在的临床异常起病较隐匿,症状不典型,容易被忽略。垂体和鞍区肿瘤术后及放疗后可并发下丘脑-垂体损伤,损伤多导致垂体前叶功能减退,同时合并垂体后叶减退症者少[31]。垂体后叶损伤引起的尿崩症通常在术后会恢复,小部分发展为永久性尿崩症(常见于术中垂体柄断裂或垂体柄肿瘤病人)。本文3例病人术后出现中枢性尿崩症。其中1例病人考虑为暂时性尿崩症,可能因术中牵拉、刮碰垂体柄及垂体后叶导致释放抗利尿激素的神经元暂时功能障碍引起。当释放抗利尿激素的神经元功能恢复时,短暂性尿崩症通常会消失[32]。在垂体前叶功能减退时,HH发生较早,但由于起病症状隐匿不典型,因此就诊时间偏晚。此5例病人均以视力下降就诊,前期性腺功能减退症状未在意或者漏诊,提示内分泌科医生和泌尿男科医生需充分重视HH的症状,减少误诊、漏诊。此5例病人均同时合并继发性肾上腺功能减退症、继发性甲状腺功能减退症,给予相应靶腺激素替代治疗,改善了病人的性功能和糖脂代谢水平,提高了病人的生活质量。
综上所述,随着基因检测和CT、MRI等影像学诊疗技术的进步,AHH的检出率逐年提高;需要尽早找出导致HH的病因,早诊断并积极治疗原发病,同时制定个体化靶腺激素替代治疗方案,提高病人的生活质量。