
2019年全球新药研发报告(Ⅰ)
2020-06-27 05:04科睿唯安
药学进展 2020年5期
( 科睿唯安 )
2019年,在全球范围内共有56种新分子实体和生物药首次获批上市(见表1)。此外,有24项重要的新产品拓展(即已上市药物的新联合用药、新制剂和新适应证等)在全球范围内获得推广。另有26种新产品,包括化学新药、生物制剂以及新产品拓展获批,但无法确认这些产品是否在2019年12月31日前上市。
研发最为活跃的治疗类药物仍为抗肿瘤药物,有13种新产品获批,其次是中枢神经系统(CNS)疾病治疗药物,有8种。
2019年有9种首创新药首次上市,其中包括治疗阿尔茨海默病(AD)、血友病和性欲减退的新药,以及数款首创的恶性肿瘤治疗药物。
2019年,美国再次成为最活跃的新药市场,其所上市的新药占全球新上市产品的56%。美国FDA一直致力于加快新药审批的进程。2017年,美国研究人员发现,2011—2015年间FDA的审批时间比欧洲药品管理局(EMA)平均缩短60 d[1];此后,美国监管机构的审批速度进一步加快。FDA正在采用快速通道和加速审批的方式批准更多的新药,而被拒绝的药物数量越来越少,这导致有人指责FDA已成为被监管行业的合作伙伴。同样值得注意的是,中国本土制药行业涌现出的化学新药和生物制剂的数量在持续增加。在2019年首次上市的产品中有7个来自中国,占全球总量的12%。
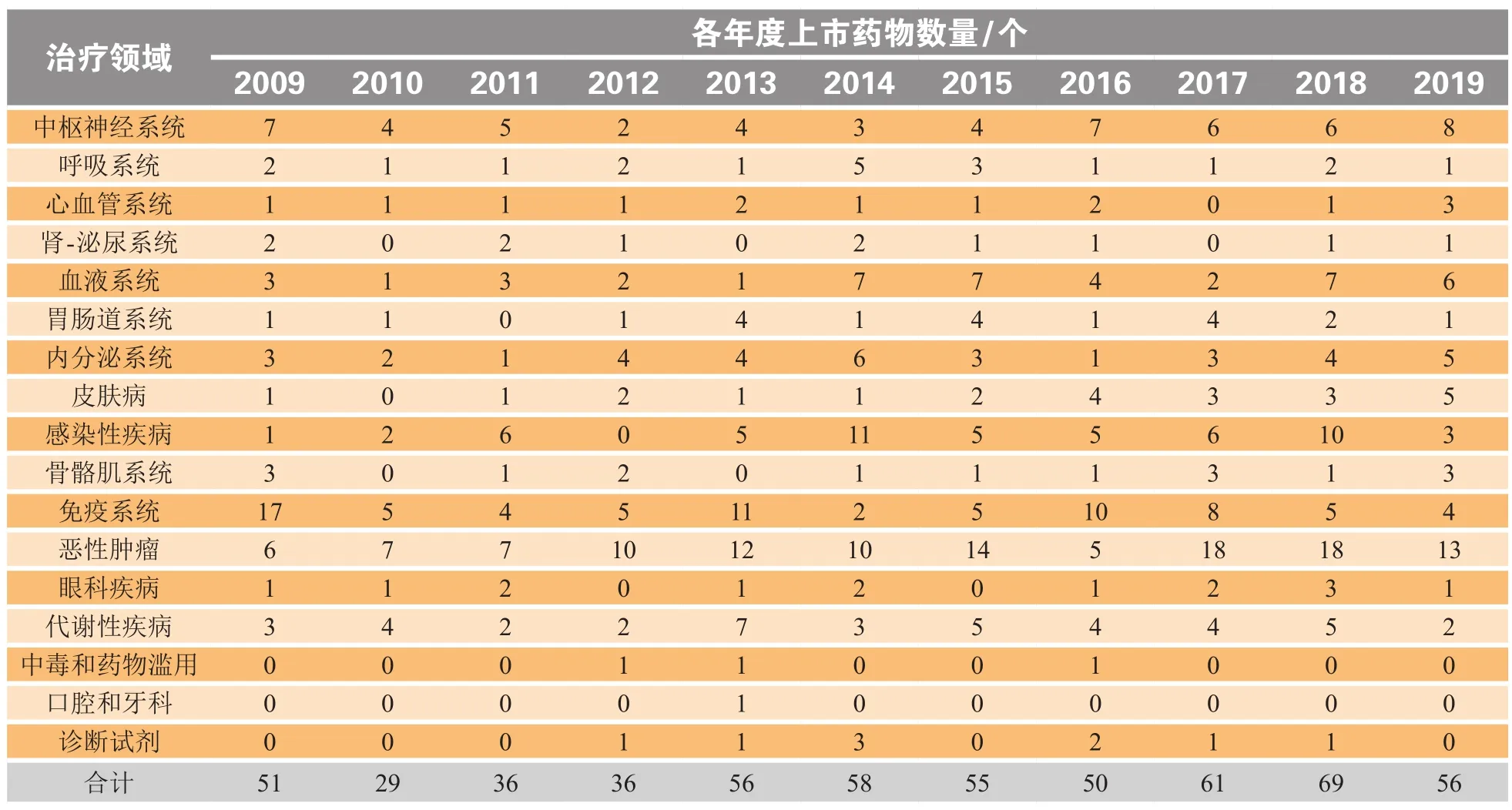
表1 2009—2019年上市的新药和生物制剂分类*Table I New drugs & biologics launched in 2009—2019 by therapeutic category
监管机构(主要为FDA,尽管其他市场也设立了相应的程序)可加快新药的开发和审批进程,并通过更多的特殊资格认证为制药公司提供激励。美国国会授权的第1个此类项目是1983年出台的孤儿药法案,该法案的构思和实施旨在促进对罕见病疗法的研究。紧随这项举措之后的是1988年实施的“快速通道”资格认定程序,旨在促进和加速药物开发和审批的进程,以治疗严重疾病并解决未满足的医疗需求。在1992年,通过了处方药申报者付费法案(PDUFA)的修订,其中包括“加速审批”和“优先审评”程序(而且,并非偶然地首次要求制药公司向监管机构支付审评费用)。1997年,PDUFA法案将目标审评时间从1年缩短至10个月。2012年,美国国会增加了“突破性疗法”的资格认定,这使得FDA能够对现有疗法有实质性改善的新药免除正常的审批程序和要求。美国批准的新药中有3/4获得某种类型的加速审评资格[2]。在欧盟,优先审评药物(PRIME)资格的实施现已进入第3个年头,其重点关注的药物是对现有疗法的疗效具有显著改善或可为无药可用的患者带来获益的药物。 2014年3月至2016年8月,欧盟药品管理局(EMA)开展了一项试点项目,以评估适应路径的方法,该方法是一种药物开发和数据生成的科学概念,在高医疗需求的疾病领域,允许早期和进展期的患者有机会更快使用新药。EMA还实施了“加速评估”项目,将未满足医疗需求的药物的审评时间从210 d缩短到150 d。在日本,Sakigake资格认定系统于2015年建立,旨在促进创新药物、医疗器械和再生药物的开发。多年来,许多国家纷纷效仿美国孤儿药项目的实施,也设立了类似的激励项目。
从这些加速审评项目中获益最大的2类疾病为恶性肿瘤和罕见病。The Wall Street Journal开展的一项研究发现,2015—2018年获批的大多数恶性肿瘤治疗药物均获得了快速通道资格认定,其中仅有19%在获批时提供了证据,表明其具有显著延长总生存期的疗效[2]。而监管机构要求的上市后研究结果,与加速审评时提供的小样本研究结果并不总是相符,因为这些小样本研究往往采用的是替代终点[3]。
总体而言,2019年在全球范围内上市的所有新药、生物制剂和产品拓展中,约有半数在上市国家被授予至少1项特殊资格认定,如表2所示。
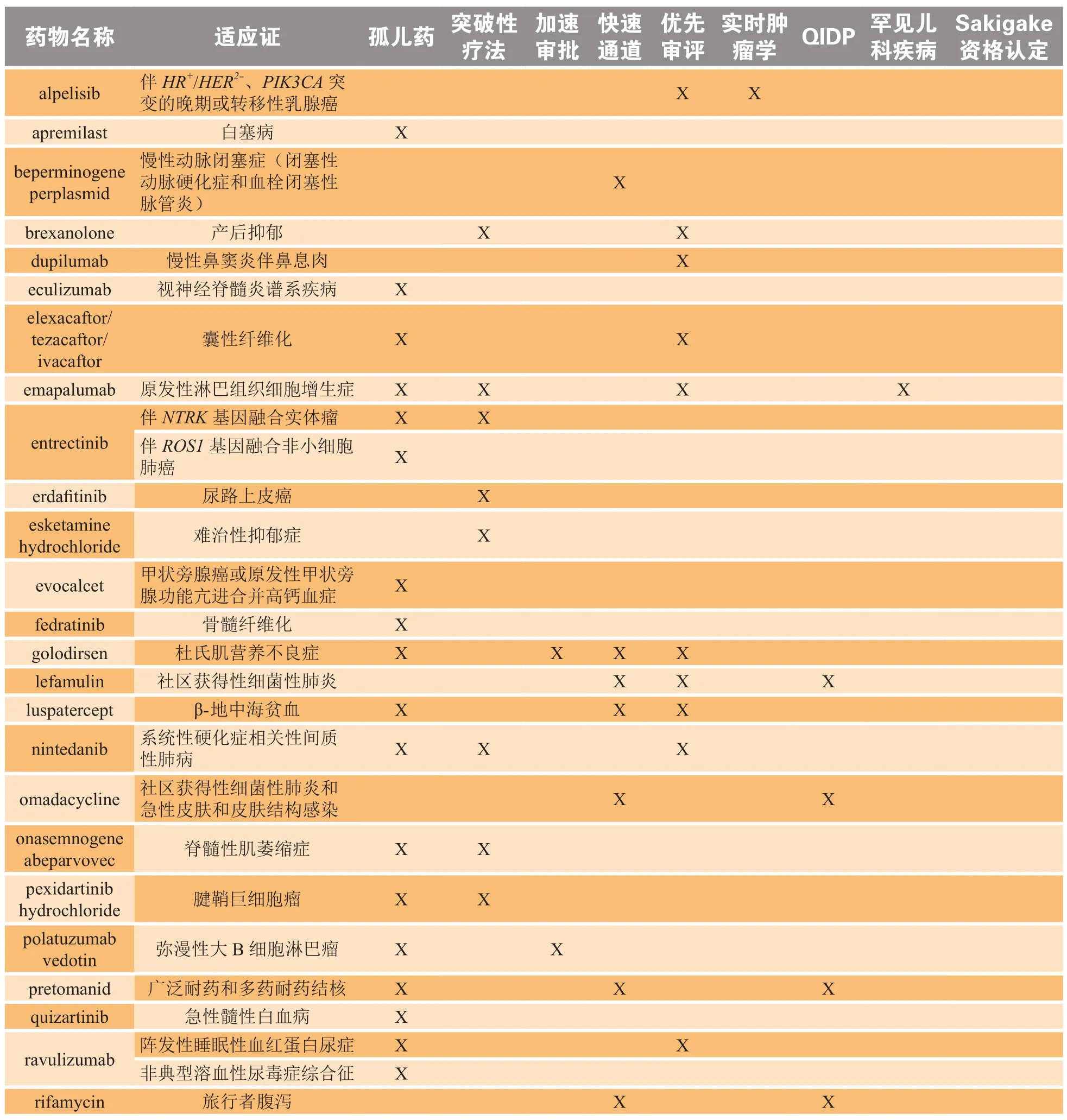
表2 特殊时期的药物开发:授予2019年新上市产品的特殊监管状态/资格认定*Table 2 Drug development in an age of exceptionality: special regulatory status/designations granted to 2019 newly launched products
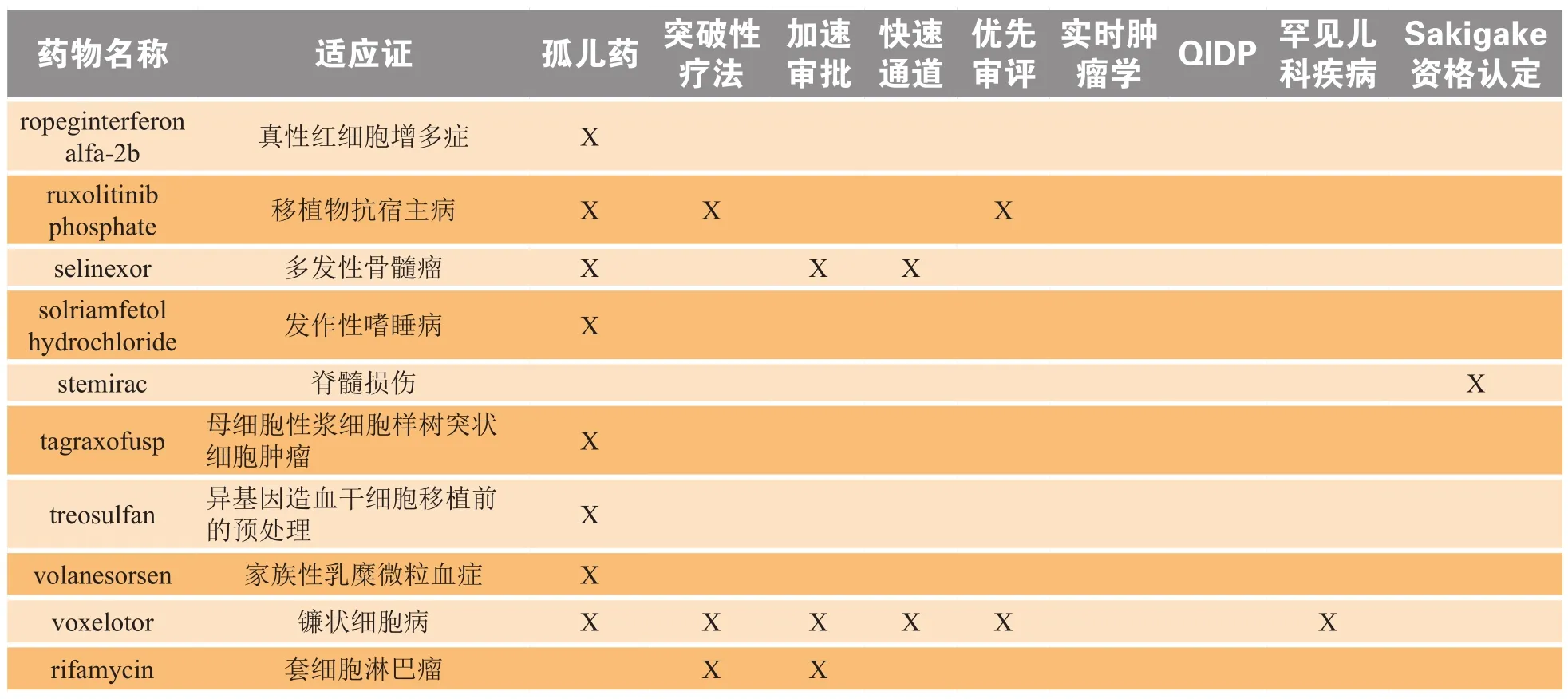
续表2
1 镇痛剂和麻醉剂
神经性疼痛是无法用单一病因或解剖学病变解释的一组异质性慢性疾病。它的发生涉及多种病因和潜在机制;典型的神经病理性疼痛综合征包括带状疱疹后遗神经痛、糖尿病神经病变、三叉神经痛、中枢性疼痛综合征、幻肢痛和格林-巴利综合征。所有神经性疼痛均以神经过度兴奋为特征,但疼痛类型和程度可能有所不同,具体取决于患者的整体健康状况和潜在的神经系统疾病等多种因素。2019年,电压依赖性钙通道亚基a-2/δ-1配体mirogabalin besylate (Tarlige;日本第一三共)在日本获批上市,用于治疗周围神经痛(PNP),包括糖尿病性PNP和带状疱疹后遗神经痛。
Lasmiditan hydrochloride (Reyvow; 礼 来)是一种口服5-HT1F受体激动剂,属于一种新型神经作用型抗偏头痛药物(NAAMA)。前几代抗偏头痛药物的作用机制均涉及血管收缩作用,而NAAMA类药物则可以在不引起血管收缩的前提下发挥疗效。Lasmiditan可选择性地靶向作用于三叉神经通路中的5-HT1F受体。2019年10月,该药被美国FDA批准用于成人偏头痛(伴或不伴先兆症状)急性治疗。礼来公司预计,该药在经过美国管制药品监督管制局(DEA)审评后,将于2020年初上市。
正如2018年全球新药和生物制剂研发报告中重点关注的那样[4],降钙素基因相关肽(CGRP)是一个颇具前景的抗偏头痛药物的新靶标。CGRP是一个由37个氨基酸组成的血管舒张神经肽,广泛分布于中枢和外周神经系统以及心血管系统,其在这些系统中发挥一系列生物学效应和生理功能,这对偏头痛的病理生理机制(包括神经调节和血管舒张)起着至关重要的作用。2018年推出了3款抗CGRP受体单克隆抗体(MAb),适应证均为偏头痛的预防性治疗,给药途径为注射。2019年,FDA批准了首款口服CGRP受体小分子抑制剂:ubrogepant(Ubrelvy),其适应证为成人偏头痛(伴或不伴先兆症状)的急性治疗。Ubrogepant由默克公司发现并于2015年授权艾尔建公司在全球进行开发和商业化。该药物计划于2020年上半年上市。
江苏恒瑞医药开发的甲苯磺酸瑞马唑仑(remimazolam tosylate)是一种苯二氮䓬类和γ-氨基丁酸(GABAA)BZ位点受体激动剂,于2019年12月下旬在中国获批,适应证为接受诊断性上消化道内镜检查时的全身麻醉。
2 精神药理学药物
根据世界卫生组织(WHO)的数据,全球范围内有超过3亿人(不分年龄段)受到抑郁症的困扰,相当于全球人口总数的4.4%。尽管患病率如此之高,但目前的疗法通常无效或仅部分有效,或者会引起无法耐受的副作用。数十年来,抑郁症的治疗药物始终是三环类抗抑郁药、单胺氧化酶抑制剂或神经递质再摄取抑制剂。2019年,2种具有新作用机制的新型抗抑郁药上市[5]。这2种药物均获得突破性疗法资格认定,且仅可在医生监督下使用。
N-甲基-D-天冬氨酸(NMDA)受体拮抗剂盐酸艾司氯胺酮(Spravato;杨森)的新型喷鼻剂已获得FDA批准,并于2019年在美国上市;该药与口服抗抑郁药联合使用,用于治疗成人难治性抑郁症(定义为对任何2种抗抑郁药治疗均无应答)。这是艾司氯胺酮的一个新适应证,该药此前作为静脉全身麻醉药已于1997年上市。在入组了1 700余例难治性抑郁症患者的临床试验中,艾司氯胺酮以亚麻醉剂量经鼻腔途径给药,与新上市的口服抗抑郁药联用,可缓解抑郁症状并延迟症状复发。值得注意的是,至少有一名FDA精神科药物咨询委员会成员认为该药物的疗效并不显著,该名成员因美国政府停摆而未出席本次会议[6]。此外,艾司氯胺酮可引起严重的副作用,包括镇静效应、解离、自杀的意念和行为[5]。2019年12月下旬,艾司氯胺酮获得欧盟委员会(EC)批准,与选择性5-羟色胺再摄取抑制剂或5-羟色胺和去甲肾上腺素再摄取抑制剂联用,用于成人难治性重度抑郁症的治疗。
四氢孕酮是突触和突触外GABAA受体的正变构调节剂,是一种在研的潜在抗抑郁药。别孕烯醇酮(Zulresso;Sage Therapeutics)是四氢孕酮的静脉注射制剂,已于2019年在美国获批上市,这是首款也是唯一一款专门用于治疗女性产后抑郁症的药物。产后抑郁症是分娩时最常见的医学并发症,在美国,每年约有40万名女性受其困扰。别孕烯醇酮使用Ligand公司开发的半合成辅料Captisol制成,这是一种经化学修饰的环糊精,其结构设计旨在增加药物的溶解度和稳定性。该药的使用需要在已通过Zulresso风险评估和缓解策略(REMS)项目认证的治疗中心,在医务人员的监督下为患者连续静脉输注2.5 d。
近几十年来,精神分裂症的治疗取得了很大进展,尤其是通过靶向多巴胺D2受体来治疗阳性症状。但是,该疾病有一系列其他症状,这些症状可严重影响患者的生活质量,因而需要使用其他治疗策略或其他靶标。2019年,一种旨在填补这一空白的新药首次获批,即IntraCellular Therapies公司开发的lumateperone tosylate(Caplyta)。
Lumateperone通过多种系统协同作用,因此代表了一种针对多种精神神经疾病的独特的治疗和管理方法。其对血清素5-HT2A受体具有强效拮抗活性,还可与多巴胺(D1和D2)受体结合,是突触前D2受体的部分激动剂和突触后多巴胺受体的拮抗剂。此外,临床前数据表明,lumateperone作为谷氨酸能磷蛋白的间接调节剂具有独特的作用机制,其可通过哺乳动物雷帕霉素(mTOR)通路靶蛋白,增加受体D1依赖的NMDA和α-氨基-3-羟基-5-甲基-4-异唑丙酸(AMPA)活性,这种作用机制可带来强效和快速的抗抑郁效果[7]。Lumateperone在2项安慰剂对照试验中显示出疗效,证实了其与安慰剂相比在主要终点,即阳性和阴性症状量表总评分方面具有显著的统计学差异。该药将于2020年上市。
2019年3月,FDA批准了Jazz Pharmaceuticals公司开发的具有双重作用机制的多巴胺和去甲肾上腺素再摄取抑制剂solriamfetol hydrochloride(Sunosi),用于改善与发作性睡病或阻塞性睡眠呼吸暂停(OSA)相关的成人日间过度嗜睡患者的醒觉状态。FDA对Sunosi的批准是基于发作性睡病过度嗜睡(TONES)Ⅲ期临床试验数据,其中包括4项随机安慰剂对照研究,证实了Sunosi相对于安慰剂的优效性。在为期12周的临床研究中,根据患者总体印象评分表的评估结果,在分别服用75 mg和150 mg Sunosi的2个治疗组中报告整体临床状况有所改善的受试者比例分别约为68% ~ 74%和78% ~90%。第12周时,通过醒觉维持试验进行疗效评估;该试验分为5次进行,第一次试验自给药后约1 h进行,第5次试验自给药后约9 h进行。在服用150 mg solriamfetol的发作性睡病患者和服用各种剂量的OSA患者的醒觉状况较安慰剂对照组均有所改善。在发作性睡病和OSA研究人群中报告的最常见不良反应(发生率不低于5%且高于安慰剂组)为头痛、恶心、食欲减退和焦虑。根据美国DEA的最终计划决定,该药已于2019年7月上市。Solriamfetol也于2020年1月在欧洲获批用于相同的适应证。
2019年12月,FDA批准了第2种针对睡眠障碍的新型药物:双重食欲素受体拮抗剂lemborexant(Dayvigo;日本卫材),适应证为以入睡和(或)睡眠维持困难为特征的成人失眠症。食欲素神经肽信号系统在醒觉维持中发挥作用。阻断促进觉醒的神经肽食欲素A和食欲素B与食欲素受体OX1和OX2的结合被认为可抑制觉醒驱动。Lemborexant与食欲素OX1和OX2受体结合,并作为竞争性拮抗剂发挥更强有效的OX2抑制作用。Lemborexant可与OX1和OX2受体结合,并作为竞争性拮抗剂发挥更强效的OX2抑制作用。Lemborexant是继2014年suvorexant上市之后第2款上市的食欲素拮抗剂。根据DEA的计划,Lemborexant将在获批90 d内在美国上市。
3 神经系统药物
近年来,肠道微生物菌群在人类健康和疾病中的关键作用日益受到关注。微生物组定义为定植于肠道、呼吸道、皮肤和人类机体其他部位的所有共生微生物的遗传物质,已知其可作为信号传导中枢,对环境刺激与宿主遗传和免疫信号进行整合以调节宿主的代谢和免疫功能,以及协调其对感染的应答。最近,有研究表明,肠道菌群的改变通过多种作用机制在AD等神经系统疾病中发挥作用[8]。2019年11月,国家药品监督管理局(NMPA)条件性批准了治疗AD的首创新药GV-971(甘露寡糖),该药被认为通过调节肠道菌群的微生态失调发挥作用。本品为分离自褐藻的酸性线状寡糖的混合物,由上海绿谷制药公司研发,适用于中轻度AD的治疗和认知功能的改善。一项在中国轻度至中度AD患者中开展的Ⅲ期临床试验发现,GV-971治疗开始后仅4周,患者的认知功能即得到统计学意义的改善,并且在为期36周的研究全程期间,每次随访访视时均观察到持续获益。根据AD评定量表认知分量表(ADAS-Cog12),治疗组和安慰剂对照组间评分的平均差值为2.54分。GV-971安全且耐受良好。GV-971在中国获得条件的批准需要进一步评价其作用机制、安全性和疗效。一项将在美国、欧洲和亚洲开展的全球多中心Ⅲ期临床试验计划于2020年初启动,以支持该产品在全球进行法规注册,预计在5年内完成。GV-971已于2019年12月底在中国上市。
Acorda Therapeutics公司开发的Inbrija是标准抗帕金森病药物levodopa的一种新型吸入制剂,2019年已在美国上市,用于使用卡比多巴/左旋多巴治疗的帕金森病患者出现关闭期症状时的间歇性治疗。关闭期症状发作的特征是在规律服用抗帕金森病药物期间出现运动和非运动症状,这些症状通常会随着疾病进展而逐渐恶化。2019年下半年,Inbrija也获得了欧盟的批准。
Cenobamate(Xcopri)是韩国SK生物制药公司发现并开发的一种新型抗惊厥药,于2019年11月获得美国FDA批准,适应证为成人部分发作性癫痫。虽然cenobamate发挥疗效的确切作用机制尚不清楚,但认为该药物可通过抑制电压门控钠电流减少神经元重复放电。该药也是GABAA受体的正变构调节剂。预计cenobamate将在通过DEA的计划审评后于2020年第2季度上市。
第2代鞘氨醇-1-磷酸(S1P)受体调节剂siponimod fumarate (Mayzent;诺华) 于 2019年春季在美国获批并上市,用于治疗成人复发型多发性硬化症,包括临床孤立综合征、复发-缓解型多发性硬化症和活动性继发性进行性多发性硬化症(SPMS)。2019年下半年,EMA人用药品委员会(CHMP)采纳了肯定意见,将siponimod用于治疗有活动性疾病的SPMS成人患者。其表现为复发或有炎症活动的影像学特征,如钆增强的T1病变或活动性、新发或扩大的T2病变。该适应证于2020年1月在欧洲获批。
Stemirac(STR-01)是日本札幌医科大学发现并由Nipro公司开发的一种新型细胞疗法,2019年已在日本获批并上市,用于脊髓损伤的治疗。Stemirac由在自体人血清中扩增的自体人骨髓来源的间充质干细胞组成。根据日本在2015年创建的Sakigake(创新药物)资格认定系统,该细胞疗法被认定为创新药物。
脊髓性肌萎缩症(SMA)是一种罕见的常染色体隐性神经退行性疾病,主要在儿童期发病,累及脊髓和脑干的运动神经元。SMA在全球范围内均有发病,其在活产婴儿中的发病率约为1/11 000,人群携带率为1/50。约有95%的SMA亚型涉及运动神经元存活基因1(SMN1)突变。直到2年前,SMA的唯一治疗方法是支持性疗法;2017年,疾病缓解性药物nusinersen的上市从根本上改变了I型SMA的治疗现状。2019年,随着onasemnogene abeparvovec(Zolgensma,由诺华子公司AveXis开发的基于腺相关病毒载体的基因疗法)在美国获批和上市,该病治疗前景进一步得到改善。Zolgensma适用于2岁以下携带SMN1基因双等位基因突变的SMA儿童患者的治疗。该药旨在通过单次一次性静脉输注人SMN基因的功能性拷贝,持续表达SMN蛋白,从而潜在地阻止疾病进展,以此针对SMA的遗传学发病机制从根本上进行治疗。该疗法具有潜在的治愈效果,这一点被用于支持其创纪录的210万美元定价的合理性。该产品在美国获得孤儿药和突破性疗法的资格认定。
2016年,尽管外周和中枢神经系统咨询委员会提出了负面建议,但FDA仍加速批准了Sarepta Therapeutics公司的首创外显子跳跃型药物eteplirsen用于治疗特定基因突变者的杜氏肌营养不良症(DMD)[9]。3年后,尽管该咨询委员会在数月前发表了否定意见(完整的回复函),FDA仍再次批准了Sarepta公司的第2款外显子跳跃型反义药物golodirsen(Vyondys 53),适用于治疗那些已证实携带了适合53号外显子跳跃的基因突变的患者。根据FDA的数据,仅有约8%的DMD患者存在这种基因突变;因此,golodirsen被认定为孤儿药。外显子跳跃疗法是基于内部缺失的抗肌萎缩蛋白可能保留其部分功能这一发现;因此,如果被破坏了的开放阅读框可以得到恢复,则可以恢复至少具有部分功能的抗肌萎缩蛋白的生成。与eteplirsen的情况相同,golodirsen获得加速审批是基于替代终点,即在一些接受该药物治疗的患者中观察到骨骼肌中的抗肌萎缩蛋白生成量出现增加。FDA得出以下结论:Sarepta公司提交的数据表明,抗肌营养不良蛋白生成量的增加“有可能合理地预测DMD患者的临床获益,这些患者已证实携带了适合53号外显子跳跃的基因突变”[10],尽管该药物的临床获益,包括运动功能的改善尚未得到确定。FDA在作出这一决定时表示,还考虑了与该药物相关的潜在风险(感染和肾毒性),以及该疾病可危及生命和使人衰弱的特点以及缺乏可用疗法的现实状况。Sarepta宣布,将立即开始对golodirsen进行商业分销。该药物后续是否能获得批准可能取决于是否能够通过验证性试验对临床获益进行验证。
视神经脊髓炎谱系疾病(NMOSD),又称Devic病,是一种累及大脑和脊髓的慢性、复发性自身免疫性炎性疾病,以单侧或双侧视神经炎和(或)脊髓炎发作为特征。NMOSD是一种罕见病,根据美国国家罕见病组织(NORD)的数据,全球患病率为1/100 000 ~ 10/100 000[11]。该病病因尚不明确,但在大约2/3的病例中,患者存在水通道蛋白-4(AQP4-IgG)抗体以及补体介导的CNS损伤,通常采用免疫抑制剂或泼尼松治疗。2019年夏季,FDA批准了NMOSD的新型治疗药物,即Alexion公司的补体抑制剂eculizumab(Soliris)。这是该抗C5单克隆抗体药物的新适应证,该药已上市,适应证包括阵发性睡眠性血红蛋白尿症(PNH)、非典型溶血性尿毒症综合征(aHUS)和重症肌无力。Eculizumab在美国被立即用于新适应证且已获得孤儿药认定;在日本,其适应证NMOSD正在接受监管审批。
4 呼吸系统药物
变应原特异性免疫治疗是一种逐渐得到广泛应用的治疗选择,用于治疗已证实对一种或几种变应原敏感的患者。Itulazax是ALK-Abell公司推出的一种新型树花粉舌下免疫疗法(SLIT),2019年在欧盟(17个国家)获批,并首次在德国上市。舌下片剂疫苗适用于治疗由来自桦木科树木(包括桦木、赤杨、山毛桃、榛树、角树和橡树)的花粉引起的中度至重度过敏性鼻炎和(或)结膜炎,且症状不能通过药物充分控制的成人患者。与可导致不良反应且必须由医生给药的皮下免疫治疗相比,SLIT疗法耐受性良好,患者可在家中服用。
2019年12月,Glenmark Pharmaceuticals公司宣布获得澳大利亚药品管理局(TGA)对固定剂量复方制剂Ryaltris(盐酸奥洛他定/糠酸莫米松)的上市许可,该复方制剂适用于治疗12岁以上过敏性鼻炎和鼻结膜炎患者。固定剂量的鼻喷剂可单次给予抗组胺药奥洛他定和皮质类固醇糠酸莫米松。该药将在澳大利亚上市,澳大利亚是全球过敏性鼻炎指数最高的国家之一(接近20%,数据来自Seqirus)。
抗白介素-4(IL-4)受体单克隆抗体dupilumab(Dupixent;Regeneron/赛诺菲)于2017年获批用于治疗特应性皮炎,2018年获批用于治疗哮喘,第3个适应证于2019年获批并上市,即与其他药物联合治疗病情未得到控制的成人慢性鼻窦炎伴鼻息肉(CRSwNP)。在dupilumab上市前,CRSwNP的唯一治疗选择是鼻用皮质类固醇或短程全身使用皮质类固醇。美国FDA经过优先审评后,基于2项关键研究(24周SINUS-24和52周SINUS-52研究)批准了dupilumab的新适应证,该2项试验评价了dupilumab 300 mg每2周1次给药加标准治疗法糠酸莫米松喷鼻剂(MFNS)相较安慰剂注射加MFNS(ClinicalTrials.gov识别码 NCT02912468和NCT02898454)的疗效。在这些研究中,dupilumab显著改善了关键疾病指标,并达到了所有主要和次要终点:治疗减小了息肉大小,减轻了鼻窦浑浊程度和症状的严重程度,且耐受性良好。EMA在2019下半年也批准了这一新适应证。
Breztri Aerosphere(布地奈德/格隆溴铵/富马酸福莫特罗)是由阿斯利康子公司Pearl Therapeutics开发的三联疗法,2019年在日本获批上市,用于缓解慢性阻塞性肺病(COPD)的症状。该疗法通过使用基于Aerosphere递送技术的加压定量吸入器单次吸入Breztri,递送吸入性皮质类固醇布地奈德、长效毒蕈碱激动剂格隆溴铵和长效β2激动剂富马酸福莫特罗。该三联疗法越来越多地用于COPD的治疗,在日本有超过500万人患有此症,因此,Breztri Aerosphere是首个在日本获批的此类产品。该疗法在欧盟和中国的申请正在审评中;美国FDA已于2019年10月签发了一份完整的回复函。
囊性纤维化跨膜传导调节因子(CFTR)蛋白嵌入在体内多种细胞的细胞膜中,在囊性纤维化(CF)的发病机制中发挥关键作用。CFTR在胰腺、汗腺、唾液腺、肠道和生殖器官内壁被覆的上皮细胞以及气道黏膜下腺中表达水平最高,而这些恰恰是CF患者体内受累及程度最高的器官和组织。CFTR最直接的作用是作为一种受cAMP调节的氯离子通道,促进氯离子双向流动。除了充当氯离子通道本身,CFTR还充当通道调节剂,影响其他氯离子通道和位于附近的细胞膜上的钠离子通道的功能。以缺陷型CFTR蛋白为靶点的新药发现标志着CF治疗的新纪元。该类药物中的第一种药物ivacaftor(Kalydeco)于2012年上市,改变了携带某些特定CFTR突变的CF亚组患者的治疗状况。含ivacaftor的联合用药(Orkambi和Symdeko)为更广泛的CF患者人群增加了治疗选择。FDA于2019年10月批准了首个含ivacaftor的复方制剂Trikafta(elexacaftor/ivacaftor/tezacaftor)[在新药申请(NDA)提交后仅3个月获批] ,并几乎立即上市。Trikafta适用于治疗年满12岁并且携带至少1个CFTR基因F508del突变拷贝的患者(无论其是否携带第2个突变)。这意味着约有90%的CF患者适合接受该三联疗法。
2019年9月,FDA批准三重激酶[血管内皮生长因子受体(VEGFR)、成纤维细胞生长因子受体(FGFR)和血小板衍生生长因子受体(PDGFR)]抑制剂nintedanib(Ofev;勃林格殷格翰)的新适应证,即减缓系统性硬化症相关性间质性肺疾病患者肺功能下降的速度。该药是FDA批准的首个用于治疗这种罕见肺部疾病的药物,在美国的审批过程中,该药经过优先审评后同时被认定为孤儿药和突破性疗法,这一结果基于一项在576例20 ~ 79岁该疾病患者中进行的随机、双盲、安慰剂对照试验(NCT02597933)而得出。患者接受了52周的治疗,部分患者的治疗时间长达100周。疗效的主要评价指标为用力肺活量;结果显示,使用尼达尼布(nintedanib)的患者肺功能下降的程度低于安慰剂组[12]。在活性药物治疗组中观察到的总体安全性特征与该治疗的已知安全性特征一致。尼达尼布治疗组报告的最常见严重不良事件为肺炎,发生率为2.8%,安慰剂组为0.3%。报告了导致永久性治疗药物减量这一不良反应的患者比例分别为34%(尼达尼布治疗组)和4%(安慰剂组);腹泻是活性药物治疗组最常见的此类不良反应。尼达尼布于2014年获批用于特发性肺纤维化成人患者的治疗,2015年获批用于非小细胞肺癌的治疗。
5 心血管系统药物
Azurity公司的Katerzia(苯磺酸氨氯地平)已于2019年在美国获批上市。这种新型钙通道阻滞剂是第1款也是唯一一款氨氯地平口服混悬剂。它可单独使用或与其他抗高血压药和抗心绞痛药联合使用,适用于治疗成人和儿童高血压以及成人冠心病。Katerzia为年满6岁需要或偏好氨氯地平口服液剂型的儿童提供了一种安全有效的即用型口服混悬液。
Daiichi Sankyo公司的esaxerenone(Minnebro)是与Exelixis公司进行研究合作期间发现的一种非甾体类、选择性盐皮质激素受体阻滞剂,于2019年在日本上市。本品适用于治疗原发性高血压,目前仅为本类药物中的第3款上市药物。盐皮质激素参与电解质和水平衡的调节;它们影响上皮细胞和肾小管中的离子转运,导致钠潴留和钾流失。
2019年3月,AnGes公 司 的beperminogene perplasmid [编码肝细胞生长因子基因(HGF)的DNA质粒]获得日本厚生劳动福利省(MHLW)的条件性批准,用于治疗重症肢体缺血(CLI)患者。Beperminogene perplasmid是在日本获批的首个基因疗法产品,适用于改善慢性动脉闭塞症(闭塞性动脉硬化症和Buerger病)患者的溃疡程度,这些患者对标准药物治疗反应不佳,且难以进行血运重建。该药基于在日本进行的一项随机安慰剂对照Ⅲ期临床试验和研究者主导的研究的结果而获批。在该批准条件下,AnGes将对所有接受该有条件批准的药物的患者进行验证性研究,并将在5年内提交解除批准条件的申请。AnGes已授权田边三菱制药公司在日本和美国对beperminogene perplasmid进行商业化,用于治疗包括CLI在内的外周动脉疾病。田边三菱于2019年9月上市了该产品,商品名为Collategene。
6 肾-泌尿系统药物
在获得欧盟批准后将近1年,阿斯利康公司的磷酸盐结合剂环硅酸钠锆(Lokelma)于2019年初在斯堪的纳维亚首次上市。该药的适应证为高钾血症,高钾血症是一种严重疾病,特征为与心血管、肾脏和代谢疾病相关的血钾水平升高。对于患有慢性肾脏疾病(CKD)的患者和服用常见治疗心力衰竭药物例如肾素-血管紧张素-醛固酮系统(RAAS)抑制剂的患者,高钾血症的发病风险显著增加。为了防止高钾血症复发,RAAS抑制剂疗法因其可能会损害心肾功能并增加死亡风险而经常被调整剂量或停止用药。3项双盲、安慰剂对照试验和一项开放性试验的数据支持了Lokelma的上市申请,其中高钾血症患者接受了长达12个月的治疗。在这些试验中,接受Lokelma治疗的患者血钾达正常水平的中位时间为2.2 h,98%的患者血钾水平在48 h内从基线值达到正常水平。Lokelma表现出长达1年的持续的血钾水平控制效果。
7 血液系统疾病药物
2019年,首创新药缺氧诱导因子脯氨酰羟化酶(HIF-PH)抑制剂roxadustat (Airuizhuo,艾瑞卓)在中国首次上市。HIF-PH抑制剂是一类新型口服活性促红细胞生成药物,通过稳定HIF复合物和刺激促红细胞生成素的内源性生成发挥作用。Roxadustat的适应证为透析依赖性(血液透析/腹膜透析)CKD患者的贫血治疗。该药由FibroGen公司开发,并授权阿斯利康公司在中国市场进行销售。
2019年6月, EC授予Bluebird Bio公司的betibeglogene darolentivec(Zynteglo)有条件上市许可,该基因疗法适用于年满12岁的非β0/β0基因型的输血依赖性β-地中海贫血(TDT)患者;这些患者适合接受造血干细胞移植(HSC),但苦于找不到合适的人白细胞抗原(HLA)配型的HSC供体而无法接受移植术。一次性基因疗法采用编码βA-T87Q-球蛋白基因的自体CD34+细胞,对TDT的潜在遗传学病因进行纠治,使符合治疗标准的患者有可能终生摆脱输血依赖。 Betibeglogene darolentivec由EMA公司的PRIME项目开发,该药也被选为EMA的自适应路径试点项目。 EMA于2019年10月下旬批准了Zynteglo的精炼生产工艺,为公司在2020年初开始上市该药品做好了准备。
FDA于2019年11月批准luspatercept(Reblozyl;Acceleron Pharma/Celgene)的适应证,即需要定期输注红细胞(RBC)的β-地中海贫血成人患者的贫血治疗。Luspatercept是首个转化生长因子β(TGF-β)抑制性红细胞成熟剂,代表一类新型疗法,其通过调节红细胞成熟后期阶段帮助患者减轻红细胞输注负荷。该药经过优先审评后,基于一项关键性、随机、双盲、安慰剂对照的多中心Ⅲ期BELIEVE研究(NCT02604433)结果获批;该研究评价了luspatercept对于需要定期输注RBC(定义为每24周输注6 ~ 20个单位的RBC,并且在5个周期无输血期不少于35一天)的β-地中海贫血成人患者的贫血治疗的疗效。该试验的主要终点达到了具有临床意义和统计学意义的改善。在luspatercept组中,21.4%的患者在随机化后第13 ~ 24周期间达到了RBC输血负荷较基线降低不低于33%(至少降低2个单位),相比之下,安慰剂组降低了4.5%。该研究还达到了关键次要终点,包括第37 ~ 48周期间输血负荷至少降低33%(至少降低2个单位),输血负荷降低的比例在luspatercept组中为19.6%,安慰剂组为3.6%。其他有效性终点包括第13 ~ 24周和第37 ~ 48周期间输血负荷至少降低50%(至少降低2个单位)。第13 ~ 24周时,观察到luspatercept治疗组中有7.6%的患者(安慰剂组中有1.8%的患者)输血负荷降低至少50%;第37 ~ 48周时,这一比例分别为10.3%和0.9%。该药被认定为孤儿药,获批后1周内在美国上市。先天性甲型血友病的主要治疗方法是在必要时给予凝血因子替代治疗联合重组凝血因子VⅢ(rFVⅢ)以达到止血或预防出血发作的目的。现有凝血因子浓缩制剂的一个主要缺点是需要频繁(通常为每日1次)给药,因此许多患者最终需要放置中心静脉通路装置。这些器械可能导致严重不良事件,如感染和血栓形成,这是该疗法的一个严重缺陷,尤其是用于儿童患者时。因此,开发长效rFVⅢ制剂已成为一个重要目标。2019年,一种此类生物制剂已在包括美国、欧盟、加拿大和日本在内的多个国家获批,即诺和诺德公司的糖基化 rFVⅢ,turoc-tocog α pegol(Esperoct)。 该 药于第3季度在德国和瑞士市场首次上市,用于治疗和预防年满12岁的甲型血友病(先天性凝血因子FVⅢ缺乏症)患者的出血。虽然该药已于2012年在欧盟获得孤儿药认定,但在2019年5月批准上市许可时,诺和诺德要求撤销这一认定。
真性红细胞增多症(PV)是一种罕见的血液病,患者体内往往生成过量的RBC。这会导致血液变得比正常情况下更黏稠,更容易形成血凝块,并增加卒中和心肌梗死的发生风险。 EC已于2019年2月批准PharmaEssentia公司的ropeginterferonalfa-2b(Besremi)单药治疗不伴有症状性脾肿大的成人PV。 Besremi是首款也是唯一一款获批的PV治疗药物,无论患者既往是否暴露于羟基脲均可使用。该批准适用于所有28个欧盟成员国以及冰岛、挪威和列支敦士登; Besremi在欧洲的上市许可持有者为AOP Orphan Pharmaceuticals公司。该药首先在奥地利和德国上市。
镰状细胞病(SCD)是一种累及红细胞的慢性、终身遗传性血液疾病。由于基因突变, SCD患者的红细胞中含有异常类型的血红蛋白,称为镰状血红蛋白(HbS)。在低氧张力状态下,这些红细胞发生聚集并变成镰状或新月形,使其难以通过小血管。镰状红细胞还表现出一种逐渐增加的倾向,即相互黏附和黏附于血管内皮细胞。 SCD患者会发生血管阻塞性危象(VOC),当红细胞变硬、变黏和失去韧性阻塞血管时就发生这种危象,导致剧烈疼痛。镰状红细胞危象可导致器官损伤、卒中、肺部并发症和包括急性胸部综合征在内的其他不良后果,而这些可能是导致该患者群体死亡的主要原因[12]。如2019年报告[4]所示, 2018年上市了50年来的首款新型SCD疗法。 2019年批准了另外2种新的治疗选择。
SCD与慢性炎症相关,可引起细胞黏附蛋白(包括P-选择素)水平上升;这会进一步增加血管和血细胞的黏性,加速血流中血细胞的黏附和聚集。这种情况可能会引起疼痛和危及生命的VOC。诺华公司的crizanlizumab(Adakveo)是一种新型SCD靶向疗法,已在PDUFA日期前2个月,即2019年11月获得FDA批准。Crizanlizumab是一种可与P-选择素结合的单克隆抗体。该批准基于一项为期52周的随机安慰剂对照SUSTAIN研究结果的支持;该研究结果显示,与安慰剂相比,crizanlizumab可使VOC的中位年发生率显著降低45%(1.63vs2.98)。此外,患者每年的中位住院天数也减少了42%(4 dvs6.87 d)[13]。
FDA在2019年11月(远早于PDUFA日期)授予了另一种SCD抗镰变首创药物voxelotor[Oxbryta;Global Blood Therapeutics(GBT) 公司]加速审批资格。Voxelotor是一种血红蛋白聚合反应抑制剂,可直接发挥抗镰变作用,已经在动物模型以及SCD患者中证实其具有疗效。Voxelotor通过增加血红蛋白与氧气的亲和力,防止镰状血红蛋白发生聚合反应以及红细胞镰变。该药的批准基于一项Ⅲ期研究HOPE(NCT03036813)的数据,该研究入组了274例年满12岁的SCD患者。研究显示,治疗24周后,voxelotor治疗组中有51%的患者达到血红蛋白升高大于 1 g · dL-1的终点(替代终点),而安慰剂组中有6.5%的患者达到血红蛋白升高大于 1 g · dL-1[14]。但是,该研究未发现VOC发生率降低。Voxelotor治疗组至少10%的患者中发生的最常见不良反应(与安慰剂组相比发生率差值大于 3%)为头痛(26%vs22%)、腹泻(20%vs10%)、腹痛(19%vs13%)、恶心(17%vs10%)、疲乏(14%vs10%)、皮疹(14%vs10%)和发热(12%vs7%)。作为加速审批的一个条件,GBT公司将在HOPE-KIDS 2研究中继续获批后的验证性研究,旨在采用经颅多普勒血流速度证明其可降低2 ~ 15岁儿童卒中的发生风险。该产品已于2019年12月上市。
2019年初,抗补体5(C5)单克隆抗体ravulizumab(Ultomiris;Alexion)在美国上市,用于治疗成人PNH患者。这种极其罕见的导致人体衰弱的血液疾病以溶血为特征,即红细胞被补体系统所破坏。PNH在所有种族、背景和年龄的男性和女性中均可发病,平均发病年龄在30岁左右。症状包括疲劳、吞咽困难、呼吸急促、腹痛、勃起功能障碍、排深色尿和贫血。慢性溶血的最严重后果是血
猜你喜欢
中国合理用药探索(2022年1期)2022-11-26
华人时刊(2022年1期)2022-04-26
昆明医科大学学报(2022年2期)2022-03-29
中老年保健(2021年3期)2021-12-03
特别健康·下半月(2019年6期)2019-08-01
祝您健康(2019年3期)2019-03-22
时代英语·高一(2018年5期)2018-11-19
时代英语·高一(2017年5期)2017-11-14
医学研究杂志(2015年2期)2015-06-10
——第一部分:新药和生物制品(Ⅲ)
药学进展(2015年4期)2015-02-10
