
烟酰胺单核苷酸对阿尔茨海默病模型大鼠认知功能的影响
2020-06-23 06:38王晓楠张励徐茜杨旸胡雪君
中国医科大学学报 2020年6期
王晓楠,张励,徐茜,杨旸,胡雪君
(中国医科大学附属第一医院老年医学科,沈阳 110001)
阿尔茨海默病 (Alzheimer disease,AD) 是以渐进性记忆和认知功能受损为主要临床症状的一种神经退行性疾病。目前,AD的病因和发病机制尚不清楚,但在AD发病机制的诸多学说中,神经细胞外β淀粉样蛋白 (β-amyloid,Aβ) 沉积处于主导地位[1]。AD的认知功能障碍主要与神经细胞能量代谢受损和氧化应激 (活性氧产生) 等密切相关[2-3]。烟酰胺腺嘌呤二核苷酸 (nicotinamide adenine dinucleotide,NAD+) 是氧化还原的辅酶,广泛参与细胞内能量代谢、信号转导、DNA修复等多种生物学效应[4]。NAD+的前体物质烟酰胺单核苷酸 (nicotinamide mononucleotide,NMN) 可增加细胞内NAD+水平。本研究采用AD大鼠模型,观察NMN对认知功能及海马CA1区锥体神经细胞的影响,以期为AD的防治提供理论依据。
1 材料与方法
1.1 材料
清洁级雄性Wistar大鼠36只,体质量280~300 g (辽宁长生生物技术股份有限公司提供)。大鼠饲养于清洁级环境中,光照、通风良好,温度、湿度适宜,进食、饮水自由。适应性饲养1周后,经Morris水迷宫测试反应正常,随机分为假手术组、AD组和AD+NMN组,每组l2只。NMN和Aβ1-42均购自美国Sigma公司。
1.2 方法
1.2.1 AD动物模型建立及干预方法:给予大鼠3.6%水合氯醛 (1 mL/100 g) 腹腔注射麻醉。将大鼠头部固定于脑立体定向仪,保持前后囟同一水平,切开硬脑膜,选取海马定位点,用微量注射器从脑表面垂直进针,将1 μL Aβ1-42 (4 μg/μL) 在5 min内缓慢注入,留针5 min,使Aβ1-42充分弥散,缓慢拔针,封补针孔,皮肤切口处用青霉素抗感染[5]。假手术组给予等体积的人工脑脊液。术后1 d大鼠完全清醒后,AD+NMN组每天给予腹膜注射NMN (500 mg/kg)干预治疗,假手术组及AD组给予腹膜注射等渗生理盐水注射液 (104.5 mg/kg),连续注射6周[6]。
1.2.2 认知功能评价:末次给药1 d后,采用Morris水迷宫评价大鼠认知功能[7]。用染发剂将每只大鼠头颈部毛发涂黑,进行定位航行;测试时室温为 (24±1)℃,将用热水溶解的奶粉1 kg加入水迷宫中,加水使安全平台低于水面1 cm,水温维持在22 ℃。每次分别从4个不同象限进行实验,将大鼠面朝池壁轻放入水中,记录潜伏期 (从大鼠进入水迷宫到爬上平台时间)、诱导次数 (如果大鼠120 s内找不到平台,可将其放回平台) 等指标。连续5 d行定位航行实验,4次/d,每次间隔20 min,于第6天撤除平台,进行空间探索实验。每次3组平行进行。
1.2.3 HE染色:常规取材、脱水、石蜡包埋,利用HE染色法观察海马CA1区神经细胞形态学改变[5]。
1.2.4 海马神经细胞内NAD+水平的测定:取出完整海马组织并切片 (厚度400 μm),放入一个微孔的膜插件内,解剖显微镜下调整海马组织切片分布。将海马组织切片捣碎溶解于75%乙醇-0.05 mol/L K2HPO4溶液,使NAD+在乙醇脱氢酶作用下转换成NADH,13 000 r/min、4 ℃离心15 min,取上清液,在400~600 nm分光光度计下测定NADH荧光值[7]。
1.3 统计学分析
采用SPSS 17.0统计软件进行统计学分析。计量资料以表示,采用单因素方差分析及Scheffe法进行多组间均数的比较,P< 0.05为差异有统计学意义。
2 结果
2.1 一般情况观察
AD组大鼠逐渐出现行动和反应迟钝,饮食减少,体质量下降,一般状态差;而AD+NMN组上述情况均较AD组大鼠有改善。
2.2 各组大鼠定位航行实验结果比较
定位航行实验结果显示,各组大鼠逃避潜伏期随时间推移逐渐递减。与假手术组比较,AD组逃避潜伏期显著延长,差异有统计学意义 (P< 0.05);与AD组比较,AD+NMN组逃避潜伏期显著缩短,差异有统计学意义 (P< 0.05)。见表1。
2.3 各组大鼠探索实验结果比较
探索实验结果显示,与假手术组比较,AD组象限活动时间及穿越次数显著减少,差异有统计学意义 (P< 0.05);与AD组比较,AD+NMN组象限活动时间及穿越次数明显增多,差异有统计学意义 (P<0.05)。见表2。
2.4 各组大鼠海马神经细胞损伤程度比较
表1 各组大鼠定位航行实验逃避潜伏期比较 (,s)Tab.1 Escape latency in each group (,s)
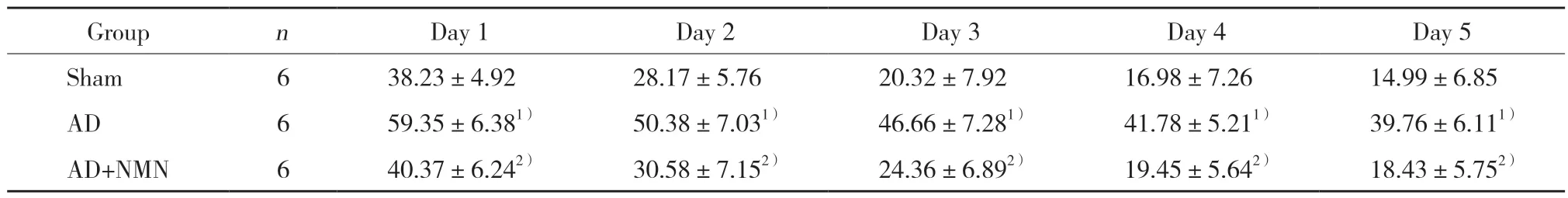
表1 各组大鼠定位航行实验逃避潜伏期比较 (,s)Tab.1 Escape latency in each group (,s)
1) P < 0.05 vs sham group;2) P < 0.05 vs AD group.
表2 各组大鼠空间探索实验象限活动时间和穿越次数比较 ()Tab.2 Time and number of crossing over a position platform ()
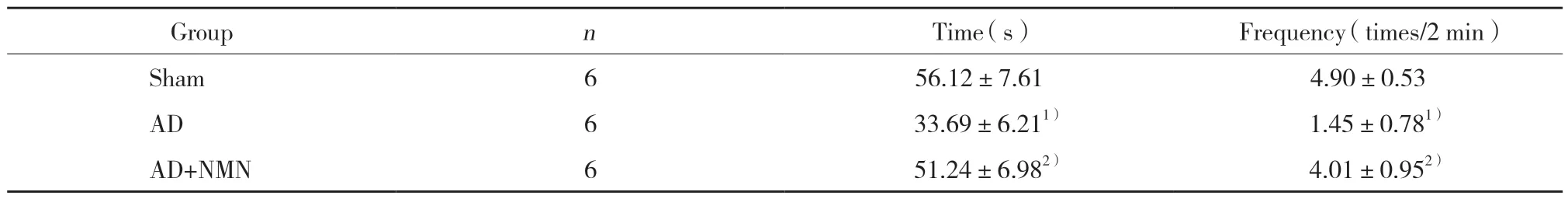
表2 各组大鼠空间探索实验象限活动时间和穿越次数比较 ()Tab.2 Time and number of crossing over a position platform ()
1) P < 0.05 vs sham group;2) P < 0.05 vs AD group.
假手术组大鼠海马CA1区锥体细胞形态正常;AD组大鼠海马CA1区锥体细胞体积肿胀或缩小,排列紊乱,细胞质致密,嗜酸性染色增强,白质区组织疏松;AD+NMN组大鼠海马CA1区锥体细胞形态正常。见图1。
2.5 海马神经细胞内NAD+水平

图1 各组大鼠海马CA1区神经细胞HE染色 ×400Fig.1 HE staining in CA1 region of rats hippocampus ×400
与假手术组[(8.52±0.85) nmol/mg protein]比较,AD组大鼠海马神经细胞内NAD+水平[(6.23±0.62)nmol/mg protein]显著下降 (P< 0.05),而与AD+NMN组[(8.47±0.56) nmol/mg protein]则无统计学差异(P> 0.05)。与AD组比较,AD+NMN组大鼠海马神经细胞内NAD+水平显著升高,差异有统计学意义 (P<0.05)。
3 讨论
研究[1]发现,AD的发病机制与神经细胞外Aβ沉积有关。可溶性Aβ寡聚体在AD出现神经变性前的早期记忆功能障碍中发挥了重要的作用[5]。Aβ寡聚体可导致神经细胞功能紊乱,是致AD记忆损伤的上游因素,主要与氧化应激、神经细胞能量受损、线粒体功能障碍以及突触传递功能抑制等密切相关[3,7]。本研究通过向大鼠海马组织内注射Aβ1-42寡聚体制作了AD动物模型。
烟酰胺磷酸核糖转移酶是细胞内NAD+合成的限速酶,可调控NAD+水平。神经细胞可以通过烟酰胺磷酸核糖转移酶调节NAD+的生物合成,从而维持神经细胞的正常形态及功能[8]。在DNA损伤时,多聚二磷酸腺苷核糖聚合酶1活性可被激活,通过水解NAD+及消耗ATP促进DNA的修复。当细胞处于氧化应激状态时,多聚二磷酸腺苷核糖聚合酶1可被大量的活性氧激活,从而过度消耗ATP和NAD+,导致神经细胞死亡,促进AD发生[9-10]。NMN是NAD+的前体,而NAD+是电子转移链中线粒体复合体Ⅰ的底物。NMN还可抑制多聚二磷酸腺苷核糖聚合酶1活性,增加细胞内NAD+水平,从而改善多聚二磷酸腺苷核糖聚合酶1介导的神经细胞死亡[11]。另一方面,NMN还可以通过直接抑制活性氧产生,发挥抗氧化应激作用[12]。
本研究中,AD组大鼠学习记忆水平明显下降,表现为逃避潜伏期显著延长,穿越次数显著减少,象限活动时间显著缩短;海马CA1区锥体细胞死亡增加;神经细胞内NAD+水平明显降低。给予NMN干预的AD+NMN组大鼠学习记忆水平较AD组明显改善,表现为逃避潜伏期明显缩短,穿越次数明显增加,象限活动时间显著延长;海马CA1区锥体细胞死亡减少;神经细胞内NAD+水平显著升高,说明NMN有效地改善了AD模型大鼠的认知功能。
综上所述,NMN可改善Aβ寡聚体诱导的海马神经细胞死亡,从而改善学习和记忆功能的损害,延缓AD进展。推测NMN可能通过抑制多聚二磷酸腺苷核糖聚合酶1活性,增加细胞内ATP生成,补充细胞内NAD+水平,发挥其神经保护作用,抗氧化作用可能是NMN神经保护作用的上游机制,仍有待进一步研究证实。
猜你喜欢
现代食品科技(2022年8期)2022-09-02
中学生数理化·高一版(2022年3期)2022-04-05
家庭医药(2021年13期)2021-12-03
昆明医科大学学报(2021年10期)2021-12-02
安徽化工(2021年5期)2021-09-07
家庭医药(2021年7期)2021-07-23
昆明医科大学学报(2021年2期)2021-03-29
初中生学习指导·提升版(2020年10期)2020-09-10
中国化妆品(2020年2期)2020-04-13
中国中医急症(2019年10期)2019-05-21
