
MnO2/C复合纳米纤维纱线的制备及其导电性能
2020-06-17 07:30:54付文丽权震震张弘楠覃小红
东华大学学报(自然科学版) 2020年2期
付文丽,权震震,b,张弘楠,b,张 坤,覃小红,b
(东华大学 a. 纺织学院; b. 纺织面料技术教育部重点实验室,上海 201620)
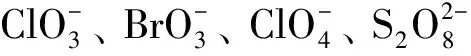
本文采用笔者课题组自主搭建的静电纺丝装置,调节纺丝参数[9]制备连续的聚丙烯腈(PAN)纳米纤维纱线原丝,经碳化得到碳纳米纤维纱线。采用易于控制的水热合成法,以KMnO4为锰源合成线状α型MnO2,并以PAN基碳纳米纱线为纳米MnO2的生长基底,通过调整反应物中的盐酸浓度合成不同形貌的MnO2/C复合纳米纤维纱线,并探究不同形貌的MnO2/C复合纳米纤维纱线的电学性能差异。
1 试验部分
1.1 试验设备及材料
试验设备:YB 302型电子天平,03-2型恒温磁力搅拌器,EST 804A型电子高压发生器,LSP01-1A型注射泵,GMSX-101型纳米纤维纱线机,MT 3000型扫描电子显微镜,S-4800型场发射扫描电子显微镜,SG 1400型管式电阻炉,UNI-T新型数字万用表。
试验材料:PAN粉末(平均相对分子质量为150 000),N-N二甲基甲酰胺(DMF)(分析纯,上海凌峰化学试剂有限公司)。
1.2 PAN基碳纳米纤维纱线的制备
通过文献查阅及静电纺丝预试验,观察不同质量分数的PAN纺丝液在纺纱过程中的出丝量、出丝稳定性、纺纱三角锥稳定性,将PAN纺丝液的质量分数确定为10%,按此配比称取一定量PAN粉末,溶于DMF中,常温下磁力搅拌12 h后形成均匀稳定的纺丝液。使用课题组自主设计搭建的静电纺纱机(如图1所示)进行纺纱,选择5 mL容量的注射器和内径为0.26 mm的针头,正负极针头间距离为12 cm,电压为±8.5 kV,注液速度为0.6 mL/h,导纱距离为8 cm,加捻金属圆盘转速为200 r/min。喷丝口的纺丝液在静电场作用力下被拉伸成连续的纳米纤维,并在位于正负极之间的金属圆盘旋转作用下形成一个稳定的加捻三角锥,在导纱杆的作用下牵引成纱,并卷绕收集在纱筒上,60 ℃真空烘燥8 h,再分别在700、 800、 900、 1 000 ℃下进行碳化,得到不同石墨化程度的碳纳米纤维纱线,分别记作C-700、 C-800、 C-900、 C-1000。
1.3 MnO2/C复合纳米纤维纱线的制备
1.4 测试仪器及测试方法
采用MT-3000型扫描电子显微镜(SEM)观察碳纳米纤维纱线的表观形态,采用S-4800型场发射扫描电子显微镜(FE-SEM)观察碳纳米纤维纱线表面生长的纳米MnO2的形貌。
使用UNI-T新型数字万用表测试并计算MnO2/C复合纳米纤维纱线的电导率,如式(1)所示。
(1)
式中:κ为电导率(S/cm);L为纱线长度(cm);R为纱线电阻(Ω);S为纱线横截面积(cm2)。
2 结果与讨论
2.1 不同碳化温度对碳纳米纤维纱线的影响
通过预氧化和碳化制备PAN碳纳米纤维纱线。先将PAN纳米纤维纱线放置在空气氛围中,以2 ℃/min的升温速率进行预氧化,升温至280 ℃后通入高纯氮气并保温2 h,使纱线在进入碳化阶段时处于高纯氮气氛围中。保温结束后以1 ℃/min的升温速率缓慢升温至360 ℃,以保证预氧丝分子链间进行充分的外环化;最后以5 ℃/min的升温速率分别升温至700、 800、 900、 1 000 ℃,保温2 h。
2.1.1 碳化温度对碳纳米纤维纱线形貌的影响
碳化前后的PAN纳米纤维纱线表面的SEM图像和纤维直径分布如图3所示。从图3可以看出:碳化前纱线中的PAN纳米纤维大体沿纱线轴向取向,碳化后纱线直径明显减小,纤维平均直径由366 nm 减小到228 nm;碳化后纱线表面的浮游纤维减少,纱线中的纤维更加致密,纤维取向更为明显。
这是由于在碳化过程中的预氧化阶段,PAN原丝中的氰基发生分子内环化,使原来的线性分子链转变为更为稳定的梯形分子链[10],PAN中的N、 H元素在与氧气反应后转化为H2O和NH3等以气体形式脱出,从而使得纤维和纱线直径减小。此外,碳化前对PAN原丝施加一定张力使得碳化过程中分子链沿纱线轴向取向[11],所以碳化后的纱线取向更为明显。表面杂乱的浮游纤维由于没有受到张力作用,在碳化过程中发生化学收缩而卷曲断裂,在氮气气流的作用下被剥离纱线主体,因此碳化后的纱线表面更为光洁,杂乱的浮游纤维大大减少。
不同碳化温度得到的碳纳米纤维纱线表面SEM图像如图4所示。从图4可以看出,随着碳化温度的升高,纱线表面越来越光洁,浮游纤维越来越少,纱线中纤维的致密度提高。
2.1.2 碳化温度对碳纳米纤维纱线电学性能的影响
不同碳化温度得到的碳纤维的X射线衍射(XRD)图如图5所示。在1 000 ℃以下的低温碳化过程中,以纤维中未环化部分的分子分解反应为主,在高纯氮气保护下,非碳元素以H2、 NH3、 HCN、 H2O等小分子形式被脱去,预氧丝中的梯形结构逐步发生热交联,初步形成类石墨结构的碳基面[12],当温度达到1 000 ℃时,纤维中碳含量达到90%[13];在高于1 000 ℃的高温碳化阶段,碳基面继续扩大,最终形成稳定的片层石墨结构。从图5中可以看到,4个样品的衍射图总体都呈现出无定型碳材料的“非晶鼓包”,在d002位置没有出现明显的衍射峰,说明该4种温度碳化得到的碳纤维石墨化程度都很低,呈无定形碳结构,没有表现出成型的石墨晶体结构。
碳纳米纤维纱线的碳化温度-电导率曲线如图6所示。从图6中可以看出:当碳化温度为700 ℃时,电导率极低,仅为0.074 S/cm,此时的碳纤维几乎没有石墨结构,导电性能差;随着温度的升高则电导率逐步上升,部分无定型碳逐渐向有序的石墨晶体结构转化,因为有序结构越多,碳纤维的导电性越好;当温度由900 ℃升至1 000 ℃时,电导率呈现陡增走势,说明从900 ℃到1 000 ℃是无定型碳结构开始大量转化为有序的类石墨结构的阶段。
2.2 不同盐酸浓度对纳米MnO2的影响
2.2.1 不同盐酸浓度对纳米MnO2形貌的影响
目前针对水热条件下MnO2的形成机理说法不一。Wang等[14]提出“卷曲-相转化”机理,即在水热环境下,锰氧八面体首先形成薄片状的δ型MnO2,当体系内没有足够的阳离子作为层状结构稳定剂时,层状结构发生卷曲生长形成纳米线,这一机理很好地解释了一维纳米MnO2的生长过程。与此类似的还有“压缩-坍塌”机理,即层状MnO2在高温、高压下被压缩,层状结构坍塌形成隧道型MnO2。此外还有“成核-溶解-各向异性生长-重结晶”机理[15],锰源在水热条件下首先生长出颗粒状的晶核,随着体系内温度和压力的上升,这些晶核逐渐团聚,并生长出各向异性的棒状结构,随着反应进行,由于MnO2生长速率较慢,生成的MnO2不足以形成棒状结构,从而由棒状晶体转变为管状。鞠广凯等[16]在探究MnO2自组装微球的形成机制时,提出H+与Cl-对管状MnO2的形成有重要作用,在压力一定的情况下,H+浓度增加会使管状MnO2的长径比增加。Zhang等[17]提出的“滚汤圆”机理中也提到在酸性条件下,层状MnO2因不稳定而发生坍塌相转化,形成2×1和1×1的隧道结构。这几种机理都表明反应物中酸的浓度对最终生成的MnO2形貌有很大影响,因此本文着重就酸浓度对MnO2形貌的影响做探究。不同盐酸浓度生成的纳米MnO2的FE-SEM图像如图7所示。
高锰酸钾是强氧化物,在加热条件下便能分解产生MnO2,在水热体系中由于加入了碳纳米纤维纱线以及盐酸,可能存在的反应有:
图7 不同盐酸浓度生成的纳米MnO2的FE-SEM图
Fig.7 FE-SEM images of nanometer MnO2generated at different molar ratio of hydrochloric acid

4MnO2+K2CO3+2KHCO3
(2)
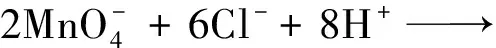
2α-MnO2+3Cl2+4H2O
(3)
当反应物中没有盐酸时,由于发生的反应主要为式(2),生成的MnO2纳米线长度较长,长度为5~10 μm,直径为20~50 nm,如图7(a)所示;当反应物中盐酸与高锰酸钾的摩尔比为4∶1时,纳米MnO2的直径为50~70 nm,长度为3~5 μm,呈短棒状,并且在头端截面处呈现管状结构,如图7(b)所示;当盐酸与高锰酸钾摩尔比提高到6∶1时,生成的MnO2纳米线仍为棒状,头端截面处的管状结构更为明显,如图7(c)所示。由此可以看出,盐酸对纳米MnO2具有一定的刻蚀作用,随着盐酸浓度的增加,刻蚀作用越明显。从外观形貌上来看,当反应物中没有盐酸时,生成的MnO2符合α型MnO2[7]的形貌特征,而当反应物中加入盐酸后,虽然产生了中空结构,但整体外观仍为具有较大长径比的线状,可以判定晶型为α型MnO2[16]。
利用红外光谱定性判定MnO2的晶体类型是一种被广泛使用的手段,高分辨率傅里叶变换红外光谱(FTIR)仪测试[18]发现,天然锰矿的红外吸收特征峰在584 cm-1处,而399、 338 cm-1处均为MnO2的红外特征吸收峰,α型MnO2在1 100 cm-1处有吸收峰。夏熙[19]在研究MnO2的红外数据时发现,斜方锰矿中含有α型MnO2和ρ型MnO2的吸收峰,但却没有显示出α型MnO2在1 100 cm-1处的特征峰。因此说明当α型MnO2与其他晶型MnO2混合时,1 100 cm-1处的吸收峰会受到干扰而不表现。不同盐酸浓度合成的MnO2的FTIR图如图8所示。由图8可以看到:3个样品在340~650 cm-1都出现了吸收峰,表明样品中均含有MnO2;而在1 100 cm-1处均没有表现出吸收峰,可能是由于有少量其他晶型MnO2干扰而不表现。当反应物中没有盐酸时,生成的MnO2在3 450和1 500 cm-1处有吸收峰,应该为O—H的伸缩振动与弯曲振动引起的[20],这表明由反应式(2)合成的α-型MnO2含有结晶水。采用液相法合成的含有结晶水的MnO2晶型通常为α-MnO2·nH2O[21]。
2.2.2 不同盐酸浓度对MnO2/C复合纳米纤维纱线的影响
图9 不同盐酸浓度合成的MnO2/C复合纳米纤维纱线表面形貌SEM图
Fig.9 SEM images of MnO2/C composite nanofiber yarns fabricated in solution of different HCl concentration
以不同温度碳化得到的碳纳米纤维纱线作为纳米MnO2的生长基底,制备得到的MnO2/C复合纳米纤维纱线的电导率也不同。试验发现,由于C-700、 C-800、 C-900本身电导率较低,其复合MnO2后,电导率均低于10-3S/cm。为了更好地比较不同盐酸浓度所得不同形貌的MnO2/C复合纳米纤维纱线的导电性能,只对电导率较高的C-1000制备得到的MnO2/C-1000复合纳米纤维纱线进行电导率性能对比。当碳纳米纤维纱线表面的MnO2厚度相同时,不同盐酸浓度合成的MnO2/C-1000复合纳米纤维纱线的电导率如表1所示。

表1 不同盐酸浓度合成的MnO2/C复合纤维纱线电导率
由表1可以看出,复合纳米纤维纱线的电导率与纯碳纳米纤维纱线相比有大幅下降。当MnO2细化到纳米级时导电性能极差,其大量地覆盖在碳纳米纤维纱线表面时阻碍了电子的传输。此外由于碳纳米纤维纱线在水热反应过程中也作为还原剂参与反应,其中的石墨结构遭到破坏,这也是导致复合纳米纤维纱线电导率大幅下降的原因。而反应物中加入不同浓度的盐酸后,尽管生成的MnO2/C-1000复合纳米纤维纱线上MnO2的晶型相同,但电导率却相差极大,可见MnO2在骨架碳纤维纱线上附着的紧密度对复合纱线的电导率有很大的影响。当盐酸与高锰酸钾浓度比为4∶1时得到的MnO2/C复合纳米纤维纱线电导率最高,为0.120 0 S/cm。
3 结 语
本文创新性地采用静电纺制备的PAN碳纳米纤维纱线作为基底,采用水热合成法,通过调整盐酸浓度,在碳纳米纤维纱线上生长不同晶体结构的MnO2纳米线,并通过红外光谱分析所得MnO2的晶型,探究了盐酸在纳米MnO2晶体生长过程中的作用。结果表明,随着盐酸浓度增加,反应体系H+浓度增加,对MnO2纳米线的刻蚀作用越强,形成管状结构越明显。复合纳米纤维纱线导电性能测试表明,盐酸浓度对MnO2在碳纳米纤维纱线表面附着的均匀程度有较大影响,从而导致复合结构的整体电导率的显著差异。
猜你喜欢
环境卫生工程(2021年4期)2021-10-13 06:52:16
纺织科学研究(2021年6期)2021-07-15 08:41:30
纺织服装流行趋势展望(2020年3期)2020-02-01 06:42:52
浙江工业大学学报(2017年5期)2018-01-22 02:03:40
现代园艺(2017年23期)2018-01-18 06:57:46
纺织服装流行趋势展望(2016年6期)2016-05-04 03:53:07
纺织服装流行趋势展望(2016年1期)2016-05-04 03:45:50
水利建设与管理(2015年10期)2015-05-09 08:29:50
技术与教育(2014年2期)2014-04-18 09:21:33
纯碱工业(2014年6期)2014-03-11 15:09:25
