
新型二氧化硅纳米毛细管开管柱的制备及其电色谱性能的研究
2020-06-07 09:34刘元元
分析科学学报 2020年2期
刘元元, 李 静, 王 彦, 闫 超*,
(1.上海交通大学药学院,上海 200240;2.上海通微分析技术有限公司,上海 201203)
开管毛细管电色谱(Open Tubular Capillary Electrochromatography,OT-CEC) 是一种新兴的在毛细管内表面涂覆或键合固定相,以电渗流为主要驱动力的色谱微分离技术[1]。它兼具了毛细管电泳(Capillary Electrophoresis,CE)和高效液相色谱(High Performance Liquid Chromatography,HPLC)双重作用机理,与HPLC相比,柱塞型的电渗流代替压力流,可以抑制样品扩散、提高柱效,而且其开放型的分离通道可以避免液相色谱柱堵塞、重复性差等问题;与CE相比,由于毛细管柱表面色谱固定相的存在,可以发挥液相色谱的高选择性优势,同时还可以抑制样品的吸附,尤其是在生物大分子或碱性物质(带正电)的分离分析中具有极好的应用前景。
固定相是制备毛细管电色谱开管柱的核心和瓶颈。SiO2纳米材料,尤其是有序介孔SiO2纳米材料(Mesoporous Silica Nanoparticles,MS NPs),不仅具有化学稳定性好、易于修饰、孔径分布窄、孔结构均一可调等特点[2,3],而且具有出色的比表面积(>1 600 m2/g)[4],可以用来接枝各种功能性基团或配体,如氨基[5]、巯基、烷基和环糊精[6],近年来作为色谱材料被广泛地用于气相色谱[7]、液相色谱[4,8,9]、离子色谱[10]、亲和色谱[11,12]以及电色谱[13]等各个领域。例如,Tian等[14]利用合成的亚微米SBA-15制备电色谱填充柱,应用于苯类同系物、苯胺类化合物以及碱性药物的分离,柱效达210 000N/m。Dong等[15]利用纤维素修饰的基于MCM-41的开管柱,可以成功应用于安息香类化合物的手性分离。
本文设计并合成了一种基于纤维状介孔二氧化硅纳米(fSiO2)和聚合物(DMAEMA polymer)的材料(P-fSiO2),将该材料通过物理吸附的方式涂覆到毛细管内壁,制备了新型的毛细管电色谱开管柱(P-fSiO2开管柱)。在考察电渗流等条件的基础上,并将其首次应用于磺胺吡啶(SP)等8种磺胺类碱性物质(图1)的开管毛细管电色谱(OT-CEC)分离分析,以考察其电色谱性能。磺胺类抗生素大多含有磺酰胺基和氨基等碱性基团,目前多采用反相高效液相色谱等[16]分离方法,而OT-CEC分离分析磺胺类化合物的方法在国内外还未见报道。

图1 8种磺胺的化学结构式Fig.1 Structural formula of 8 sulfonamides
1 实验部分
1.1 仪器、试剂和材料
S-4800场发射扫描电子显微镜(SEM)(日本,Hitachi公司);JEOL-2100F场发射透射电子显微镜(TEM)(日本,电子株式会社);Sartorius BT224S电子天平(北京赛多利斯仪器系统有限公司);KQ5200DE型超声波清洗(昆山市超声仪器有限公司);D2F-1B真空干燥箱(上海博泰实验设备有限公司);DF-101S集热式恒温磁力搅拌器(河南巩义市予华仪器有限责任公司);DM36-12马弗炉(上海德恭实业有限公司);电阻丝加热器(苏州环球色谱有限责任公司)。
磺胺吡啶(SP)、磺胺二甲基嘧啶(SM2)、磺胺二甲氧嘧啶(SDM)、磺胺喹恶啉(SQX)、磺胺甲氧哒嗪(SMP)、磺胺间甲氧嘧啶(SMM)、磺胺二甲异恶唑(SIZ)、磺胺嘧啶(SD)8种标准品购自阿拉丁试剂(上海)有限公司。磺胺标准溶液的配制:分别准确称取磺胺吡啶、磺胺二甲基嘧啶、磺胺二甲氧嘧啶、磺胺喹恶啉、磺胺甲氧吡啶嗪、磺胺间甲氧嘧啶、磺胺二甲异恶唑、磺胺嘧啶各10.0 mg,加入色谱纯乙腈定容至10.0 mL,配成浓度为1.0 mg/mL的标准储备溶液,用时取适量混合再稀释到所需浓度,过0.22 μm滤膜,并置于4 ℃避光保存。无水乙醇、丙酮、尿素、环己烷、异丙醇、硼砂、Na2HPO4、NaOH、HCl、十六烷基三甲基溴化铵(CTAB)、正硅酸四乙酯(TEOS)等均购买于国药集团化学试剂有限公司(上海);无水二氯甲烷、CuCl2、CuCl、2-溴异丁酰溴、3-氨基丙基三乙氧基硅烷(APTES)、2,2′-联吡啶(BPY)、4-二甲氨基吡啶(DMAP)、甲基丙烯酸二甲基氨基乙酯(DMAEMA)等均购自阿拉丁试剂(上海)有限公司;甲醇、乙腈(色谱纯)购自Tedia公司。熔融石英毛细管(50 μm i.d.,363 μm o.d.)购自河北永年锐沣色谱器件有限公司。纯水由Milli-Q纯化系统(美国,Millipore)制备。
1.2 P-fSiO2的合成
首先合成纤维状介孔二氧化硅纳米材料(fSiO2NPs)。在装有磁力搅拌器、回流冷凝管、温度计和滴液漏斗的250 mL四颈瓶中,加入0.5 g(1.3 mmol)CTAB、0.3 g(5.0 mmol)尿素和15 mL纯水,开动搅拌使其溶解后,分别加入15 mL环己烷和0.46 mL(6.0 mmol)异丙醇,随后缓慢滴加1.25 g(6.0 mmol)TEOS并剧烈搅拌30 min,加热使溶液温度达到72 ℃,反应10 h。反应结束后,将反应液冷却至室温,以5 000 r/min离心3 min,用纯水和丙酮分别洗涤3次,60 ℃干燥24 h。最后将合成的固体转移至50 mL 0.6%NH4NO3-乙醇溶液中,60 ℃回流4 h除去模板剂CTAB。反应结束后,冷却至室温,用乙醇洗涤3次,60 ℃干燥24 h,得到白色纤维状介孔二氧化硅纳米材料(fSiO2)。
通过原子转移自由基聚合(Atom Transfer Radical Polymerization,ATRP)法将DMAEMA接枝到除去模板的fSiO2上。将1.0 gfSiO2加入到含有0.06 mol/L APTES的 50 mL无水乙腈中,25 ℃反应24 h。反应结束后,以5 000 r/min离心3 min,乙腈洗涤3次,60 ℃干燥得到氨基化fSiO2(NH2-fSiO2)。将1.0 g NH2-fSiO2加入到含有0.15 mol/L三乙胺,0.13 mol/L 2-溴异丁酰溴以及催化量DMAP的50 mL无水二氯甲烷中,25 ℃反应12 h。结束后,以5 000 r/min离心3 min,二氯甲烷洗涤3次,60 ℃干燥得到溴化-氨基化f-SiO2(Br-NH2-f-SiO2)。最后,将1.0 g Br-NH2-fSiO2加入到10 mL甲醇-水(2∶1,V/V)溶液中,超声溶解后,加入0.5 mol/L DMAEMA,0.08 mol/L BPY,0.006 mol/L CuCl2和0.02 mol/L CuCl,室温反应12 h后,用纯水洗涤至无色,60 ℃干燥得到DMAEMA-fSiO2(P-fSiO2)。
P-fSiO2的合成路线如图2所示。
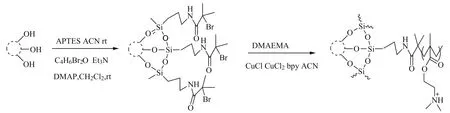
图2 P-fSiO2合成示意图Fig.2 Synthesis DMAEMA-modified of silica nanoparticles
1.3 P-fSiO2开管柱的制备
毛细管预处理:将总长度65 cm,有效长度45 cm,50 μm i.d.的熔融石英毛细管依次用甲醇(30 min)、纯水(5 min)、0.1 mol/L NaOH溶液(60 min)、纯水(5 min)、0.1 mol/L HCl(30 min)、纯水(5 min)、甲醇(30 min)冲洗,以充分暴露毛细管内壁硅羟基,80 ℃干燥过夜后备用。P-fSiO2开管柱的制备:P-fSiO2开管柱的制备原理主要是利用P-fSiO2与负电性的石英毛细管壁之间的静电吸附(Electrostatic Attraction)作用。过程如下:2.0%P-fSiO2乙醇溶液以5.0 μL/min灌入活化干燥后的毛细管,持续5 min,70 ℃干燥12 h后,用纯水洗涤5 min,70 ℃干燥6 h得到P-fSiO2开管柱,备用。
1.4 毛细管电色谱条件
CE-1000毛细管电泳仪(Unimicro(Shanghai)Technologies,Shanghai,China),由毛细管柱上检测器、高压电源及色谱工作站(Trisep-2003)组成;色谱柱:P-fSiO2开管柱(50 μm i.d,有效长度45 cm,总长度65 cm);流动相:硼砂体系;进样量:电动进样10 kV、5 s;检测波长:254 nm;温度:25 ℃。
2 结果与讨论
2.1 色谱材料和开管柱的表征
透射电子显微镜(TEM)显示合成的P-fSiO2纳米材料为350 nm,如图3a所示。等温吸脱附线是研究多孔材料表面和孔的基本数据。如图3b所示,根据IUPAC分类的6种吸脱附等温线,该材料的N2吸附-脱附等温线属于“Ⅳ”型,脱附等温线在吸附等温线的上方,根据介孔回滞环的类型,为H3型迟滞环,迟滞环的存在说明材料堆积形成了介孔。对其比表面积的分析一般采用Brunauer-Emmett-Teller(BET)方法,比表面积是544.56 m2/g,孔径分布通常采用Barrett-Joiner-Halenda(BJH)模型,计算得到的平均孔径分布为3.41 nm。

图3 P-fSiO2的透射电镜(TEM)图(a)和fSiO2的N2吸附-脱附等温线(b)Fig.3 TEM image of P-fSiO2(a) and nitrogen adsorption-desorption isotherm of fSiO2(b)
2.2 P-fSiO2开管柱的表征
扫描电子显微镜(SEM)是表征毛细管内壁形态的有效方法,图4a以及其局部放大图4c显示,空管的内壁形态光滑、无涂覆迹象;而P-fSiO2开管柱的SEM图(图4b、4d),显示有一层明显的颗粒状固定相的存在,局部放大(图4d)显示填料的粒径大小同图3a。

图4 空管(a)、P-fSiO2开管柱(b)的扫描电镜(SEM)以及局部放大(c、d)图Fig.4 SEM images of bare capillary(a),P-fSiO2 column(b) and magnified SEM(c,d)
2.3 P-fSiO2开管电色谱柱的性能评价
2.3.1 电渗流的考察在电色谱技术中,电渗流(EOF)作为流动相的主要驱动力,其大小直接影响色谱分离效果。本实验以二甲基亚砜(DMSO)为电渗流标记物,如图5所示,P-fSiO2开管柱的电渗流随pH值的降低而减小,且在相应pH点的电渗流值均小于毛细管空管,尤其是在碱性pH条件下,电渗流值显著降低。因此,基于P-fSiO2涂层材料对电渗流的抑制作用,或许可以在碱性样品的分离分析中得到良好的应用,接下来本文选取磺胺类碱性化合物作为P-fSiO2开管柱性能考察的标的物。

图5 缓冲液pH对空管和P-fSiO2开管柱的电渗流的影响Fig.5 Effect of buffer pH on EOF of bare capillary and P-fSiO2 open-tubular columnconditions:bare capillary and P-fSiO2 column(50 μm i.d.,65/45);running buffer:30 mmol/L sodium borate,pH=4.0 - 9.0;voltage:-18 kV;temperature:25 ℃;sample,DMSO;detection:220 nm.
2.3.2 缓冲盐及浓度的考察在考察电渗流的大小及方向的基础上,本文首先考察了磷酸盐和硼砂缓冲溶液等不同体系的影响。结果表明,在磷酸盐缓冲体系中,基线不稳定;且同等浓度的缓冲盐,硼砂的离子强度小,有利于避免较大焦耳热的产生。综合考虑,选择硼砂缓冲溶液作为运行缓冲液。随着硼砂缓冲盐浓度的增加,双电层厚度增加,电渗流降低,带电溶质在开管柱内的迁移速率变慢,同时较大的离子强度增大了焦耳热,不仅影响基线的稳定而且影响实验的重复性。实验考察了不同浓度缓冲溶液(25~40 mmol/L)的影响(图6)。当缓冲液浓度为40 mmol/L时,缓冲液离子强度大,表观电流显著增大,焦耳热增大,溶质扩散加剧,峰形展宽;当使用较低浓度缓冲液(25 mmol/L)时,迁移时间虽然较快,但峰高响应有所降低,检出灵敏度有所下降。综合考虑,实验优化的硼砂缓冲液浓度为30 mmol/L。
2.3.3 分离电压的考察分离电压主要影响分析时间与分离效果两个方面。在30 mmol/L硼砂缓冲体系下,经过实验考察(图7),当分离电压低于16 kV时,分离度增加,但分析时间稍长。当电压高于20 kV时,分析时间缩短,但分离度变差,并且此时由于焦耳热效应明显,基线波动较大,使整个检测体系较不稳定。因此,综合分离度、分析时间和缓冲体系稳定性等多方面的影响,实验选择的分离电压为16~20 kV之间。

图6 缓冲液浓度对P-fSiO2开管柱分离效果的影响Fig.6 Effect of buffer concentration on the seperation efficiency of OTCEC on P-fSiO2 open-tubular columnconditions:P-fSiO2 column(50 μm i.d.,65/45);running buffer:25 - 40 mmol/L sodium borate,pH=9.0;voltage:-15 kV;temperature:25 ℃;detection:254 nm.peak identification:1.SP;2.SM2;3.SDM;4.SQX;5.SMP;6.SMM;7.SIZ;8.SD.

图7 电压对P-fSiO2开管柱分离效果的影响Fig.7 Effect of high voltage on the seperation efficiency of OTCEC on P-fSiO2 open-tubular columnconditions:P-fSiO2 column(50 μm i.d.,65/45);running buffer:30 mmol/L sodium borate,pH 9.0;voltage:12 - 20 kV;temperature:25 ℃;detection:254 nm;peak identification.1.SP;2.SM2;3.SDM;4.SQX;5.SMP;6.SMM;7.SIZ;8.SD.
2.3.4 缓冲液pH的考察在不同的pH环境中,因分子解离程度的不同会造成待测物的电泳速率的差异,因此缓冲液的pH是分离过程中的重要考察因素。磺胺类化合物含有磺酰胺基和氨基等碱性基团,大部分pKa在5~8之间(pKa:SP,8.4;SM2,6.05;SDM,6.8;SQX,5.5;SMP,7.2;SMM,6.05;SIZ,4.83;SD,6.5)。在优化的条件(30 mmol/L硼砂缓冲液,16 kV)和预实验基础上,本文选取了7.0、8.0以及9.0三个点考察pH对磺胺分离的影响。如图8所示,与pH=8.0的缓冲液相比,当缓冲液pH=7.0时,虽然峰1与8的分离度变大,但是分离时间稍长(~60 min),且峰4~6的分离度变差,峰7出现了前移的现象;当缓冲液pH=9.0时,分离时间虽有所缩短,但峰1~8整体的分离度变小。因此,实验选取缓冲液的较佳pH条件为8.0。
2.3.5 与空管比较在优化的分离条件下(30 mmol/L硼砂缓冲液,18 kV,pH=8.0),实验比较了P-fSiO2开管柱与空管的分离能力。如图9所示,没有涂层的毛细管柱(即电泳模式)虽然出峰较快(时间为9~11 min),但很难实现对8种磺胺的分离;而同样的实验条件下,P-fSiO2开管柱(即电色谱模式)由于涂层材料的存在,不仅可以实现对8种磺胺的良好分离(出峰时间为14~31 min),而且SQX的柱效高达228 542N/m(表1)。我们推测,在P-fSiO2开管电色谱分离模式中,胺基聚合物材料的存在,对磺胺可能展现了一定的氢键和疏水性作用。

表1 8种磺胺的柱效数据
2.3.6 重复性在相同条件下,将同一根柱子在1 d内连续进样6次,计算出峰时间的日内精密度(RSD%,n=6);同一根柱子连续6 d,每天进样6次,计算出峰时间的日间RSD(%,n=6);同时选取5根柱子以标记物的出峰时间计算柱间RSD(%,n=5),结果如表2所示。8种磺胺的日内RSD值小于4.3%,日间RSD小于4.9%,柱间RSD为2.2%。说明,P-fSiO2开管柱具有良好的稳定性,且对磺胺类物质的分离具有良好的重现性。

图8 缓冲液pH对P-fSiO2开管柱分离效果的影响Fig.8 Effect of buffer pH on the seperation efficiency of OTCEC on P-fSiO2 open-tubular columnconditions:P-fSiO2 column(50 μm i.d.,65/45);running buffer:30 mmol/L sodium borate,pH 7.0 - 9.0;voltage:-16 kV;temperature:25 ℃;detection:254 nm;peak identification.1.SP;2.SM2;3.SDM;4.SQX;5.SMP;6.SMM;7.SIZ;8.SD.

图9 磺胺类化合物在P-fSiO2开管柱与空管中的分离效果Fig.9 Separation of sulfmides in P-fSiO2 open-tubular column and bare capillaryconditions:bare capillary and P-fSiO2 column(50 μm i.d.,65/45);running buffer:30 mmol/L sodium borate,pH=8.0;voltage:-18 kV;temperature:25 ℃;detection:254 nm;peak identification.1.SP;2.SM2;3.SDM;4.SQX;5.SMP;6.SMM;7.SIZ;8.SD.

表2 P-fSiO2开管柱的日内、日间以及柱间重复性
N/A,NotApplicable.
3 结论
本文采用物理吸附的方式,将合成的P-fSiO2材料涂覆到毛细管内壁,制备了一种新型的毛细管电色谱P-fSiO2开管柱,并将其应用于磺胺类物质的OT-CEC分离分析以验证其电色谱性能。P-fSiO2开管柱的制备方法简便、快捷、涂层稳定。通过对缓冲溶液浓度、pH值以及分离电压等因素的考察,可以实现对8种磺胺类化合物的分离,且重复性好。因此,基于P-fSiO2柱的OT-CEC方法有望在磺胺类等碱性物质的分离分析中得到良好的应用。
猜你喜欢
世界最新医学信息文摘(2022年17期)2022-07-27
文萃报·周五版(2022年24期)2022-06-21
西南石油大学学报(自然科学版)(2019年4期)2019-11-04
华东师范大学学报(自然科学版)(2019年2期)2019-06-11
奥秘(创新大赛)(2019年2期)2019-03-07
北方人(2018年16期)2018-08-20
爱你(2016年4期)2016-12-06
大自然探索(2015年12期)2015-09-10
云南畜牧兽医(2015年4期)2015-02-28
中国兽药杂志(2011年10期)2011-10-30
